![]()
Condrodisplasia Puntata
Condrodisplasia Puntata
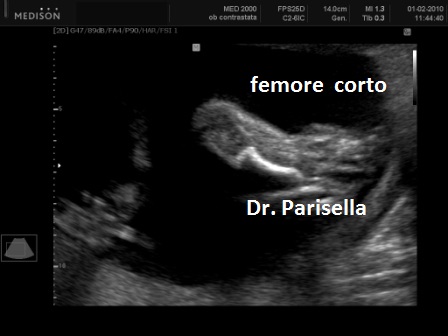
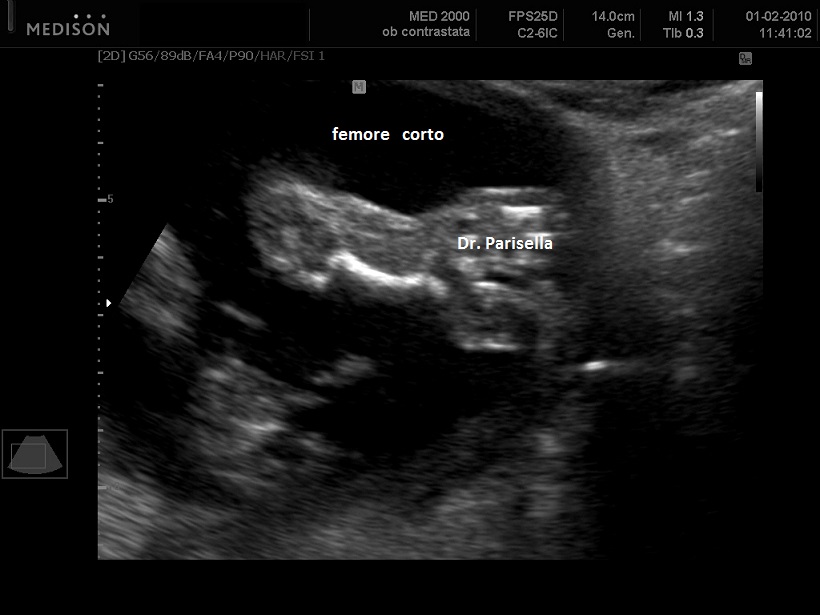
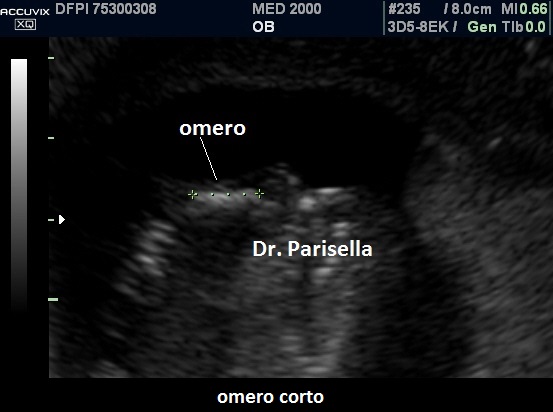
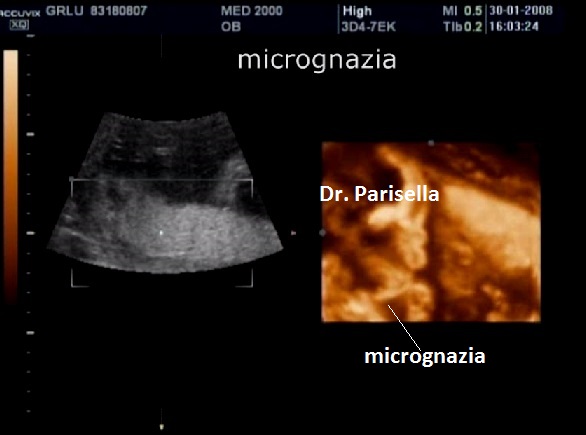
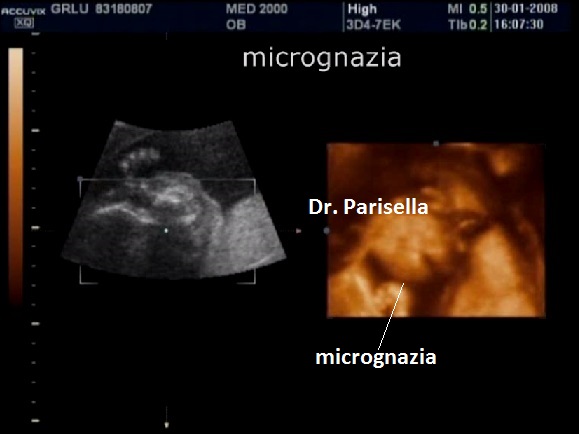
La condrodisplasia puntata rizomelica ha una prevalenza stimata in 1:100.000. E' caratterizzata da nanismo che colpisce soprattutto il tratto rizomelico ( femore e omero ), tipico aspetto del viso (ampio ponte nasale, epicanto, palato ogivale, orecchie esterne displasiche, micrognazia) alterazioni vertebrali, cataratta, lesioni cutanee e grave ritardo mentale. Le alterazioni vertebrali consistono caratteristicamente in lesioni coronali dei corpivertebrali. La malattia è causata da un difetto del metabolismo dei perossisomi e viene trasmessa come carattere autosomico recessivo.
Ne esistono tre forme:
Condrodisplasia Puntata Rizomelica Tipo I o RCDP1 OMIM 215100
Condrodisplasia Puntata Rizomelica Tipo II o RCDP2 OMIM 222765
Condrodisplasia Puntata Rizomelica Tipo III o RCDP3 OMIM 600121
La diagnosi si basa sul quadro clinico e radiografico e può essere confermata dall'analisi molecolare.
La diagnosi prenatale è possibile quando la mutazione genica è già stata identificata nella famiglia. Non è disponibile una specifica terapia per la correzione del difetto enzimatico. La condrodisplasia rizomelica ha una prognosi sfavorevole e la morte di solito sopravviene nella prima decade di vita, in particolare per le complicazioni respiratorie.
CONDRODISPLASIA PUNTATA TIPO NON RIZOMELICO
Questo gruppo comprende varie forme a trasmissione diversa e prevalenza non nota, che sono:
Condrodisplasia Puntata legata all'X dominante o Sindrome di Conradi-Hunermann OMIM 302960
Condrodisplasia Puntata Brachitelefalangica OMIM 302950
Condrodisplasia Puntata tibio-metacarpale OMIM 118651
Condrodisplasia Puntata di Sheffield OMIM 118650
Sindrome di Toriello OMIM 215105
La Condrodisplasia Puntata legata all'X Dominante o Sindrome di Conradi-Hünermann può essere diagnosticata in epoca prenatale, di solito nel III trimestre. E' caratterizzata da bassa statura legata ad ipoplasia di omero e femore (l'omero è colpito principalmente), asimmetria degli arti, scoliosi, epifisi slargate con multipli foci iperecogeni, contratture articolari; segni presenti ma non evidenziabili all'ecografia sono l'eritroderma ittiosiforme lamellare e la cataratta, che può essere monolaterale. L'intelligenza è normale. La malattia colpisce prevalentemente le femmine ed è più grave o addirittura letale nei maschi. È trasmessa come carattere dominante legato all'X, dovuta a mutazioni nel gene EBP che codifica per un enzima coinvolto nel metabolismo del colesterolo.
La condrodisplasia brachitelefalangica si associa a gravi dismorfismi facciali caratterizzati da ipoplasia della parte mediana della faccia, con naso abnormemente corto e sella appiattita, mascella iposviluppata, prognatismo e/o 'morso inverso' (o malocclusione di classe III), calcificazioni soprattutto nel tarso e negli arti inferiori e falangi distali ipoplasiche. La statura e l'intelligenza sono normali o pressoché normali. La malattia è ereditata come carattere recessivo legato all'X ed è causata da una mutazione nel gene ARSE.
L'assunzione materna di anticoagulanti in gravidanza, il deficit dei fattori della coagulazione dipendenti dalla vitamina K e il deficit materno di vitamina K possono causare sintomi simili a quelli della condrodisplasia puntata brachitelefalangica. Le calcificazioni in prossimità delle epifisi possono essere presenti anche nella sindrome feto-alcolica e nei feti nati da madri con lupus eritematoso sistemico.
Condrodiplasia puntata di tipo tibio-metacarpale: presenta facies tipica (piatta, naso piccolo), arti corti (tibie piccole, II e III osso metacarpale piccoli), schisi coronale dei corpi vertebrali, punteggature calcifiche.
Condrodisplasia puntata di tipo Sheffield: è una forma lieve, autosomica dominante,caratterizzata da facies tipica, calcificazioni puntiformi del calcagno, statura bassa.
Sindrome di Toriello: in aggiunta alle epifisi slargate con multipli foci iperecogeni è caratterizzata da bassa statura, lievi anomalie facciali e coloboma.
La condrodisplasia puntata può essere diagnosticata ecograficamente in epoca prenatale, di solito negli stadi avanzati della gravidanza, ma la classificazione esatta richiede approfondimenti biochimici (ricerca di steroli anomali e acidi grassi a catena molto lunga) sui campioni di liquido amniotico. La prognosi è molto variabile.
Bibliografia
Agematsu, K., Koike, K., Morosawa, H., Nakahori, Y.,
Nakagome, Y., Akabane, T. Chondrodysplasia punctata with X;Y translocation.
Hum. Genet. 80: 105-107, 1988.
Allansmith, M., Senz, E. H. Chondrodystrophia congenita punctata
(Conradi's disease). Am. J. Dis. Child. 100: 109-116, 1960. Asanti, R., Heikel,
P.-E. Chondroangiopathia calcarea or punctata. Ann. Paediat. Fenn. 9: 280-289,
1963.
Ausavarat, S., Tanpaiboon, P., Tongkobpetch, S.,
Suphapeetiporn, K., Shotelersuk, V. Two novel EBP mutations in
Conradi-Hunermann-Happle syndrome. Europ. J. Derm. 18: 391-393, 2008.
Austin-Ward, E., Castillo, S., Cuchacovich, M., Espinoza,
A., Cofre-Beca, J., Gonzalez, S, Solivelles, X., Bloomfield, J. Neonatal lupus
syndrome: a case with chondrodysplasia punctata and other unusual
manifestations. J. Med. Genet. 35: 695-697, 1998.
Ballabio, A., Bardoni, B., Carrozzo, R., Andria, G., Bick,
D., Campbell, L., Hamel, B., Ferguson-Smith, M. A., Gimelli, G., Fraccaro, M.,
Maraschio, P., Zuffardi, O., Guioli, S., Camerino, G. Contiguous gene syndromes
due to deletions in the distal short arm of the human X chromosome. Proc. Nat.
Acad. Sci. 86: 10001-10005, 1989.
Ballabio, A., Zollo, M., Carrozzo, R., Caiulo, A., Zuffardi,
O., Cascioli, C. F., Viggiano, D., Strisciuglio, P. Deletion of the distal
short arm of the X chromosome (Xp) in a patient with short stature,
chondrodysplasia punctata, and X-linked ichthyosis due to steroid sulfatase deficiency.
Am. J. Med. Genet. 41: 184-187, 1991.
Barr, D. G. D., Kirk, J. M., Al Howasi, M., Wanders, R. J.
A., Schutgens, R. B. H. Rhizomelic chondrodysplasia punctata with isolated
DHAP-AT deficiency. Arch. Dis. Child. 68: 415-417, 1993.
Becker, M. H., Genieser, N. B., Finegold, M., Miranda, D.,
Spackman, T. Chondrodysplasia punctata: is maternal warfarin therapy a factor?
Am. J. Dis. Child. 129: 356-359, 1975.
Bergstrom, K., Gustavson, K.-H., Jorulf, H.
Chondrodystrophia calcificans congenita (Conradi's disease) in a mother and her
child. Clin. Genet. 3: 158-161, 1972.
Bick, D., Curry, C. J. R., McGill, J. R., Schorderet, D. F.,
Bux, R. C., Moore, C. M. Male infant with ichthyosis, Kallmann syndrome,
chondrodysplasia punctata, and an Xp chromosome deletion. Am. J. Med. Genet.
33: 100-107, 1989.
Bodian, E. L. Skin manifestations of Conradi's disease:
chondrodystrophia congenita punctata. Arch. Derm. 94: 743-748, 1966.
Borochowitz, Z. Generalized chondrodysplasia punctata with
shortness of humeri and brachymetacarpy: humero-metacarpal (HM) type: variation
or heterogeneity? Am. J. Med. Genet. 41: 417-422, 1991.]
Braverman, N., Lin, P., Moebius, F. F., Obie, C., Moser, A.,
Glossmann, H., Wilcox, W. R., Rimoin, D. L., Smith, M., Kratz, L., Kelley, R.
I., Valle, D. Mutations in the gene encoding
3-beta-hydroxysteroid-delta(8),delta(7)-isomerase cause X-linked dominant
Conradi-Hunermann syndrome. Nature Genet. 22: 291-294, 1999.
Braverman, N., Steel, G., Obie, C., Moser, A., Moser, H.,
Gould, S. J., Valle, D. Human PEX7 encodes the peroxisomal PTS2 receptor and is
responsible for rhizomelic chondrodysplasia punctata. Nature Genet. 15:
369-376, 1997.
Brites, P., Motley, A. M., Gressens, P., Mooyer, P. A. W.,
Ploegaert, I., Everts, V., Evrard, P., Carmeliet, P., Dewerchin, M.,
Schoonjans, L., Duran, M., Waterham, H. R., Wanders, R. J. A., Baes, M.
Impaired neuronal migration and endochondral ossification in Pex7 knockout
mice: a model for rhizomelic chondrodysplasia punctata. Hum. Molec. Genet. 12:
2255-2267, 2003.
Brookhyser, K. M., Lipson, M. H., Moser, A. B., Moser, H.
W., Lachman, R. S., Rimoin, D. L. Prenatal diagnosis of rhizomelic
chondrodysplasia punctata due to isolated alkyldihydroacetonephosphate
acyltransferase synthase deficiency. Prenatal Diag. 19: 383-385, 1999.
Bruch, D., Megahed, M., Majewski, F., Ruzicka, T. Ichthyotic
and psoriasiform skin lesions along Blaschko's lines in a woman with X-linked
dominant chondrodysplasia punctata. J. Am. Acad. Derm. 33: 356-360, 1995.
Brunetti-Pierri, N., Andreucci, M. V., Tuzzi, R., Vega, G.
R., Gray, G., McKeown, C., Ballabio, A., Andria, G., Meroni, G., Parenti, G.
X-linked recessive chondrodysplasia punctata: spectrum of arylsulfatase E gene
mutations and expanded clinical variability. Am. J. Med. Genet. 117A: 164-168,
2003.
Burck, U. Mesomelic dysplasia with punctata epiphyseal
calcifications--a new entity of chondrodysplasia punctata? Europ. J. Pediat.
138: 67-72, 1982
Burck, U., Schaefer, E., Held, K. R. Mesomelic dysplasia
with short ulna, long fibula, brachymetacarpy and micrognathia: clinical and
radiological differential diagnostic features. Pediat. Radiol. 9: 161-165,
1980.
Ciske, D. J., Waggoner, D. J., Dowton, S. B. Unique cardiac
and cerebral anomalies with chondrodysplasia punctata. Am. J. Med. Genet. 75:
59-61, 1998.
Clayton, P. T., Eckhardt, S., Wilson, J., Hall, C. M.,
Yousuf, Y., Wanders, R. J. A., Schutgens, R. B. H. Isolated
dihydroxyacetonephosphate acyltransferase deficiency presenting with
developmental delay. J. Inherit. Metab. Dis. 17: 533-540, 1994.
Comings, D. E., Papazian, C., Schoene, H. R. Conradi's
disease (chondrodystrophia calcificans congenita, congenital stippled
epiphyses). J. Pediat. 72: 63-69, 1968.
Curry, C. J. R., Lanman, J. T., Jr., Magenis, R. E., Brown,
M. G., Bergner, E. A., Shapiro, L. J. X-linked chondrodysplasia punctata with
ichthyosis: chromosomal localization to Xp. (Abstract) Am. J. Hum. Genet. 34:
122A, 1982.
Curry, C. J. R., Magenis, R. E., Brown, M., Lanman, J. T.,
Jr., Tsai, J., O'Lague, P., Goodfellow, P., Mohandas, T., Bergner, E. A.,
Shapiro, L. J. Inherited chondrodysplasia punctata due to a deletion of the
terminal short arm of an X chromosome. New Eng. J. Med. 311: 1010-1015, 1984
De Lange, C., Janssen, T. Congenital chondrodystrophia
calcificans of infant in association with other abnormalities: case. Maandschr.
Kindergeneesk. 17: 67-74, 1949.
de Vet, E. C. J. M., IJlst, L., Oostheim, W., Wanders, R. J.
A., van den Bosch, H. Alkyl-dihydroxyacetonephosphate synthase: fate in
peroxisome biogenesis disorders and identification of the point mutation
underlying a single enzyme deficiency. J. Biol. Chem. 273: 10296-10301, 1998.
Derry, J. M. J., Gormally, E., Means, G. D., Zhao, W.,
Meindl, A., Kelley, R. I., Boyd, Y., Herman, G. E. Mutations in a
delta(8)-delta(7) sterol isomerase in the tattered mouse and X-linked dominant
chondrodysplasia punctata. Nature Genet. 22: 286-290, 1999
Dimmick, J., Applegarth, D., Chitayat, D., Clarke, L., Lau,
A., Pantzar, T., Moser, H. Rhizomelic chon 10. drodysplasia punctata in an
infant with del(4)(p14p16). (Abstract) Am. J. Hum. Genet. 49 (suppl.): 155,
1991.
Eash, D. D., Weaver, D. D., Brunetti-Pierri, N. Cervical
spine stenosis and possible vitamin K deficiency embryopathy in an unusual case
of chondrodysplasia punctata and an updated classification system. Am. J. Med.
Genet. 122A: 70-75, 2003.
Elcioglu, N., Hall, C. M. Maternal systemic lupus
erythematosus and chondrodysplasia punctata in two sibs: phenocopy or
coincidence? J. Med. Genet. 35: 690-694, 1998
Elias, E. R., Mobassaleh, M., Hajra, A. K., Moser, A. B.
Developmental delay and growth failure caused by a peroxisomal disorder,
dihydroxyacetonephosphate acyltransferase (DHAP-AT) deficiency. Am. J. Med.
Genet. 80: 223-226, 1998.
Franco, B., Meroni, G., Parenti, G., Levilliers, J.,
Bernard, L., Gebbia, M., Cox, L., Maroteaux, P., Sheffield, L., Rappold, G. A.,
Andria, G., Petit, C., Ballabio, A. A cluster of sulfatase genes on Xp22.3:
mutations in chondrodysplasia punctata (CDPX) and implications for warfarin
embryopathy. Cell 81: 1-20, 1995.
Fraser, F. C., Scriver, J. B. A hereditary factor in
chondrodystrophia calcificans congenita. New Eng. J. Med. 250: 272-277, 1954.
Fritsch, H., Manzke, H. Beitrag zur Chondrodystrophia
calcificans connata (Conradi-Hunermann-Syndrom). Arch. Kinderheilk. 169:
235-254, 1963.
Gallop, P. M., Lian, J. B., Hauschka, P. V. Carboxylated
calcium-binding proteins and vitamin K. New Eng. J. Med. 302: 1460-1466, 1980.
Gekle, D. Ein Beitrag zum Problem der Chondrodystrophia
calcificans congenita. Arch. Kinderheilk. 169: 267-273, 1963.
Gilbert, E. F., Opitz, J. M., Spranger, J. W., Langer, L.
O., Jr., Wolfson, J. J., Viseskul, C. Chondrodysplasia punctata--rhizomelic
form: pathologic and radiologic studies of three infants. Europ. J. Pediat. 12:
89-109, 1976.
Gray, R. G. F., Green, A., Chapman, S., McKeown, C.,
Schutgens, R. B. H., Wanders, R. J. A. Rhizomelic chondrodysplasia punctata--a
new clinical variant. J. Inherit. Metab. Dis. 15: 931-932, 1992.
Hall, J. G., Pauli, R. M., Wilson, K. M. Maternal and fetal
sequelae of anticoagulation during pregnancy. Am. J. Med. 68: 122-138, 1980.
Happle, R. Cataracts as a marker of genetic heterogeneity in
chondrodysplasia punctata. Clin. Genet. 19: 64-66, 1981
Happle, R. X-linked dominant chondrodysplasia punctata:
review of literature and report of a case. Hum. Genet. 53: 65-73, 1979.]
Happle, R., Matthass, H.-H., Macher, E. Sex-linked
chondrodysplasia punctata?. Clin. Genet. 11: 73-76, 1977
Happle, R., Phillips, R. J. S., Roessner, A., Junemann, G.
Homologous genes for X-linked chondrodysplasia punctata in man and mouse. Hum.
Genet. 63: 24-27, 1983.
Harrod, M. J. E., Sherrod, P. S. Warfarin embryopathy in
siblings. Obstet. Gynec. 57: 673-676, 1981.
Haynes, E. R., Wangner, W. F. Chondroangiopathia calcarea
seu punctata: review and case report. Radiology 57: 547-550, 1951.
Heikoop, J. C., van Roermund, C. W. T., Just, W. W., Ofman,
R., Schutgens, R. B. H., Heymans, H. S. A., Wanders, R. J. A., Tager, J. M.
Rhizomelic chondrodysplasia punctata: deficiency of 3-oxoacyl-Coenzyme A
thiolase in peroxisomes and impaired processing of the enzyme. J. Clin. Invest.
86: 126-130, 1990.
Heikoop, J. C., Wanders, R. J. A., Strijland, A., Purvis,
R., Schutgens, R. B. H., Tager, J. M. Genetic and biochemical heterogeneity in
patients with the rhizomelic form of chondrodysplasia punctata--a complementation
study. Hum. Genet. 89: 439-444, 1992.
Herman, G. E. Disorders of cholesterol biosynthesis:
prototypic metabolic malformation syndromes. Hum. Molec. Genet. 12(R1):
R75-R88, 2003.
Herman, G. E., Kelley, R. I., Pureza, V., Smith, D., Kopacz,
K., Pitt, J., Sutphen, R., Sheffield, L. J., Metzenberg, A. B. Characterization
of mutations in 22 females with X-linked dominant chondrodysplasia punctata
(Happle syndrome). Genet. Med. 4: 434-438, 2002.
Herman, G. E., Walton, S. J. Close linkage of the murine
locus bare patches to the X-linked visual pigment gene: implications for
mapping human X-linked dominant chondrodysplasia punctata. Genomics 7: 307-312,
1990.
Heselson, N. G., Cremin, B. J., Beighton, P. Lethal
chondrodysplasia punctata. Clin. Radiol. 29: 679-684, 1978
Heymans, H. S. A., Oorthuys, J. W. E., Nelck, G., Wanders,
R. J. A., Schutgens, R. B. H. Rhizomelic chondrodysplasia punctata: another
peroxisomal disorder. (Letter) New Eng. J. Med. 313: 187-188, 1985.
Heymans, H. S., Wanders, R. J. A. Rhizomelic
chondrodysplasia punctata.In: Moser, H. W. (ed.) : Handbook of Clinical
Neurology: Neurodystrophies and Neurolipidoses. Amsterdam: Elsevier Science
B.V. 1996. Pp. 525-533.
Ikegawa, S., Ohashi, H., Ogata, T., Honda, A., Tsukahara,
M., Kubo, T., Kimizuka, M., Shimode, M., Hasegawa, T., Nishimura, G., Nakamura,
Y. Novel and recurrent EBP mutations in X-linked dominant chondrodysplasia
punctata. Am. J. Med. Genet. 94: 300-305, 2000.
Itzkovitz, B., Jiralerspong, S., Nimmo, G., Loscalzo, M.,
Horovitz, D. D. G., Snowden, A., Moser, A., Steinberg, S., Braverman, N.
Functional characterization of novel mutations in GNPAT and AGPS, causing
rhizomelic chondrodysplasia punctata (RCDP) types 2 and 3. Hum. Mutat. 33:
189-197, 2012.
Jenkins, T., Noll, B. Chondrodysplasia punctata: report of
parent-to-child transmission. S. Afr. Med. J. 54: 22-25, 1978.
Johnson, W. G. Metabolic interference and the +/-
heterozygote: a hypothetical form of simple inheritance which is neither dominant
nor recessive. Am. J. Hum. Genet. 32: 374-386, 1980.
Josephson, B. M., Oriatti, M. D. Chondrodystrophia
calcificans congenita: report of a case and review of the literature.
Pediatrics 28: 425-435, 1961
Kalter, D. C., Atherton, D. J., Clayton, P. T. X-linked
dominant Conradi-Hunermann syndrome presenting as congenital erythroderma. J.
Am. Acad. Derm. 21: 248-256, 1989.
Kapusta, L., Brunner, H. G., Hamel, B. C. J.
Craniofrontonasal dysplasia. Europ. J. Pediat. 151: 837-841, 1992.
Kelley, R. I., Wilcox, W. G., Smith, M., Kratz, L. E.,
Moser, A., Rimoin, D. S. Abnormal sterol metabolism in patients with
Conradi-Hunermann-Happle syndrome and sporadic lethal chondrodysplasia
punctata. Am. J. Med. Genet. 83: 213-219, 1999. Note: Erratum: Am. J. Med.
Genet. 84: 387 only, 1999.
Khanna, A. J., Braverman, N. E., Valle, D., Sponseller, P.
D. Cervical stenosis secondary to rhizomelic chondrodysplasia punctata. Am. J.
Med. Genet. 99: 63-66, 2001.
Kozlowski, K., Basel, D., Beighton, P. Chondrodysplasia
punctata in siblings and maternal lupus erythematosus. Clin. Genet. 66:
545-549, 2004.
Kozlowski, K., Godlonton, J., Gardner, J., Beighton, P.
Lethal non-rhizomelic dysplasia epiphysealis punctata. Clin. Dysmorph. 11:
203-208, 2002.
Liu, X. Y., Dangel, A. W., Kelley, R. I., Zhao, W., Denny,
P., Botcherby, M., Cattanach, B., Peters, J., Hunsicker, P. R., Mallon, A.-M.,
Strivens, M. A., Bate, R., Miller, W., Rhodes, M., Brown, S. D. M., Herman, G.
E. The gene mutated in bare patches and striated mice encodes a novel
3-beta-hydroxysteroid dehydrogenase. Nature Genet. 22: 182-187, 1999.
Manzke, H., Christophers, E., Weidemann, H.-R. Dominant
sex-linked inherited chondrodysplasia punctata: a distinct type of
chondrodysplasia punctata. Clin. Genet. 17: 97-107, 1980.
Maroteaux, P. Brachytelephalangic chondrodysplasia punctata:
a possible X-linked recessive form. Hum. Genet. 82: 167-170, 1989.
Maroteaux, P. Nomenclature internationale des maladies
osseuses constitutionelles. Ann. Radiol. 13: 455-464, 1970.
Matos-Miranda, C., Nimmo, G., Williams, B., Tysoe, C.,
Owens, M., Bale, S., Braverman, N. A prospective study of brachytelephalangic
chondrodysplasia punctata: identification of arylsulfatase E mutations,
functional analysis of novel missense alleles, and determination of potential
phenocopies. Genet. Med. 15: 650-657, 2013.
Melnick, J. C. Chondrodystrophia calcificans congenita
(chondrodysplasia epiphysialis punctata, stippled epiphyses). Am. J. Dis.
Child. 110: 218-225, 1965
Menger, H., Lin, A. E., Toriello, H. V., Bernert, G.,
Spranger, J. W. Vitamin K deficiency embryopathy: a phenocopy of the warfarin
embryopathy due to a disorder of embryonic vitamin K metabolism. Am. J. Med.
Genet. 72: 129-134, 1997.
Moser, A. B., Rasmussen, M., Naidu, S., Watkins, P. A.,
McGuinness, M., Hajra, A. K., Chen, G., Raymond, G., Liu, A., Gordon, D.,
Garnaas, K., Walton, D. S., Skjedal, O. H., Guggenheim, M. A., Jackson, L. G.,
Elias, E. R., Moser, H. W. Phenotype of patients with peroxisomal disorders
subdivided into sixteen complementation groups. J. Pediat. 127: 13-22, 1995.
Motley, A. M., Hettema, E. H., Hogenhout, E. M., Brites, P.,
ten Asbroek, A. L. M. A., Wijburg, F. A., Baas, F., Heijmans, H. S., Tabak, H.
F., Wanders, R. J. A., Distel, B. Rhizomelic chondrodysplasia punctata is a
peroxisomal protein targeting disease caused by a non-functional PTS2 receptor.
Nature Genet. 15: 377-380, 1997.
Motley, A. M., Tabak, H. F., Smeitink, J. A. M., Poll-The,
B. T., Barth, P. G., Wanders, R. J. A. Non-rhizomelic and rhizomelic
chondrodysplasia punctata within a single complementation group. Biochim.
Biophys. Acta 1315: 153-158, 1996.
Nimmo, G., Monsonego, S., Descartes, M., Franklin, J.,
Steinberg, S., Braverman, N. Rhizomelic chrondrodysplasia (sic) punctate type 2
resulting from paternal isodisomy of chromosome 1. Am. J. Med. Genet. 152A:
1812-1817, 2010.
Nino, M., Matos-Miranda, C., Maeda, M., Chen, L., Allanson,
J., Armour, C., Greene, C., Kamaluddeen, M., Rita, D., Medne, L., Zackai, E.,
Mansour, S., Superti-Furga, A., Lewanda, A., Bober, M., Rosenbaum, K.,
Braverman, N. Clinical and molecular analysis of arylsulfatase E in patients
with brachytelephalangic chondrodysplasia punctata. Am. J. Med. Genet. 146A:
997-1008, 2008
Nivelon-Chevallier, A. Vitamin K deficiency embryopathy. Am.
J. Med. Genet. 79: 66-68, 1998.
Norwood, C., Stephan, M., Leston, W. Further delineation of
X-linked dominant chondrodysplasia punctata. (Abstract) Proc. Greenwood Genet.
Center 4: 130-131, 1985.
Ofman, R., Hettema, E. H., Hogenhout, E. M., Caruso, U.,
Muijsers, A. O., Wanders, R. J. A. Acyl-CoA:dihydroxyacetonephosphate
acyltransferase: cloning of the human cDNA and resolution of the molecular
basis in rhizomelic chondrodysplasia punctata type 2. Hum. Molec. Genet. 7:
847-853, 1998.
Petit, C., Melki, J., Levilliers, J., Serville, F.,
Weissenbach, J., Maroteaux, P. An interstitial deletion in Xp22.3 in a family
with X-linked recessive chondrodysplasia punctata and short stature. Hum.
Genet. 85: 247-250, 1990.
Pettifor, J. M., Benson, R. Congenital malformations
associated with the administration of oral anticoagulants during pregnancy. J.
Pediat. 86: 459-462, 1975.
Philips, L. I. Chondrodystrophia calcificans congenita. New
Zeal. Med. J. 56: 22-27, 1957.
Phillips, R. J. S., Hawkes, S. G., Moseley, H. J.
Bare-patches, a new sex-linked gene in the mouse, associated with a high
production of XO females. I. A preliminary report of breeding experiments.
Genet. Res. 22: 91-99, 1973.
Phillips, R. J. S., Kaufman, M. H. Bare-patches, a new
sex-linked gene in the mouse, associated with a high production of XO females.
II. Investigation into the nature and mechanism of the XO production. Genet.
Res. 24: 22-41, 1974.
Poll-The, B. T., Maroteaux, P., Narcy, C., Quetin, P.,
Guesnu, M., Wanders, R. J. A., Schutgens, R. B. H., Saudubray, J. M. A new type
of chondrodysplasia punctata associated with peroxisomal dysfunction. J.
Inherit. Metab. Dis. 14: 361-363, 1991.
Poulos, A., Sheffield, L., Sharp, P., Sherwood, G., Johnson,
D., Beckman, K., Fellenberg, A. J., Wraith, J. E., Chow, C. W., Usher, S.,
Singh, H. Rhizomelic chondrodysplasia punctata: clinical, pathologic, and
biochemical findings in two patients. J. Pediat. 113: 685-690, 1988.
Purdue, P. E., Zhang, J. W., Skoneczny, M., Lararow, P. B.
Rhizomelic chondrodysplasia punctata is caused by deficiency of human PEX7, a
homologue of the yeast PTS2 receptor. Nature Genet. 15: 381-384, 1997.
Putschar, W. G. J. Chondrodystrophia calcificans congenita
(dysplasia epiphysialis punctata). Bull. Hosp. Joint Dis. 12: 514-527, 1951.
Rittler, M., Menger, H., Spranger, J. Chondrodysplasia
punctata, tibia-metacarpal (MT) type. Am. J. Med. Genet. 37: 200-208, 1990
Rodemer, C., Thai, T.-P., Brugger, B., Kaercher, T., Werner,
H., Nave, K.-A., Wieland, F., Gorgas, K., Just, W. W. Inactivation of ether
lipid biosynthesis causes male infertility, defects in eye development and
optic nerve hypoplasia in mice. Hum. Molec. Genet. 12: 1881-1895, 2003.
Rosenfield, R. L., Breibart, S., Isaacs, H., Klevit, H. D.,
Mellman, W. J. Trisomy of chromosomes 13-15 and 17-18: its association with
infantile arteriosclerosis. Am. J. Med. Sci. 244: 763-779, 1962.
Ryan, S. G., Chance, P. F., Zou, C.-H., Spinner, N. B.,
Golden, J. A., Smietana, S. Epilepsy and mental retardation limited to females:
an X-linked dominant disorder with male sparing. Nature Genet. 17: 92-95, 1997.
Savarirayan, R., Boyle, R. J., Masel, J., Rogers, J. G.,
Sheffield, L. J. Longterm follow-up in chondrodysplasia punctata,
tibia-metacarpal type, demonstrating natural history. Am. J. Med. Genet. 124A:
148-157, 2004.
Sefiani, A., Heuertz, S., Turleau, C., Thibaud, D., de
Grouchy, J., Hors-Cayla, M.-C. Incontinentia pigmenti: Xp breakpoint is not the
same in a case of r(X) and in X/autosome translocations. Ann. Genet. 32:
149-151, 1989.
Seidel, J., Schiller, S., Kelbova, C., Beensen, V., Orth,
U., Vogt, S., Claussen, U., Zintl, F., Rappold, G. A. Brachytelephalangic
dwarfism due to the loss of ARSE and SHOX genes resulting from an X;Y
translocation. Clin. Genet. 59: 115-121, 2001.
Seo, K. W., Miyoshi, H., Kon, Y., Watanabe, T. Chromosomal
mapping and developmental study of Tattered-Hokkaido (Tdho). Mammalian Genome
8: 578-580, 1997
Shaul, W. L., Emery, H., Hall, J. G. Chondrodysplasia
punctata and maternal warfarin use during pregnancy. Am. J. Dis. Child. 129:
360-362, 1975.
Sheffield, L. J. Comment on Traupe's tribute to Rudolf
Happle. (Letter) Am. J. Med. Genet. 101: 283 only, 2001.
Sheffield, L. J., Danks, E. M., Mayne, V., Hutchinson, L. A.
Chondrodysplasia punctata: 23 cases of a mild and relatively common variety. J.
Pediat. 89: 916-923, 1976.
Sheffield, L. J., Halliday, J. L., Danks, D. M., Rogers, J.
G., Poulos, A., Morrison, N. Clinical, radiological and biochemical
classification of chondrodysplasia punctata. (Abstract) Am. J. Hum. Genet. 45
(suppl.): A64, 1
Sheffield, L. J., Osborn, A. H., Hutchison, W. M., Sillence,
D. O., Forrest, S. M., White, S. J., Dahl, H.-H. M. Segregation of mutations in
arylsulphatase E and correlation with the clinical presentation of
chondrodysplasia punctata. J. Med. Genet. 35: 1004-1008, 1998. [PubMed:
9863597, related citations] [Full Text: HighWire Press]
Shirahama, S., Miyahara, A., Kitoh, H., Honda, A., Kawase,
A., Yamada, K., Mabuchi, A., Kura, H., Yokoyama, Y., Tsutsumi, M., Ikeda, T.,
Tanaka, N., Nishimura, G., Ohashi, H., Ikegawa, S. Skewed X-chromosome
inactivation causes intra-familial phenotypic variation of an EBP mutation in a
family with X-linked dominant chondrodysplasia punctata. Hum. Genet. 112:
78-83, 2003
Silengo, M. C., Luzzatti, L., Silverman, F. N. Clinical and
genetic aspects of Conradi-Hunermann disease: a report of three familial cases
and review of the literature. J. Pediat. 97: 911-917, 1980.
Silverman, F. Discussion on the relation between stippled
epiphyses and the multiple forms of epiphyseal dysplasia. Birth Defects Orig.
Art. Ser. V(4): 68-70, 1969.
Silverman, F. N. Dysplasies epiphysaires: entite
proteiforme. Ann. Radiol. 4: 833-867, 1961.
Spranger, J. W., Opitz, J. M., Bibber, U. Heterogeneity of
chondrodysplasia punctata. Hum. Genet. 11: 190-212, 1971.
Stenflo, J., Suttie, J. W. Vitamin K-dependent formation of
gamma-carboxyglutamic acid. Ann. Rev. Biochem. 46: 157-172, 1977.
Sugarman, G. I. Chondrodysplasia punctata (rhizomelic type):
case report and pathologic findings.In: Bergsma, D. : Skeletal Dysplasias.
Amsterdam: Excerpta Medica (pub.) 1974. Pp. 334-340.
Sutphen, R., Amar, M. J., Kousseff, B. G., Toomey, K. E. XXY
male with X-linked dominant chondrodysplasia punctata (Happle syndrome). Am. J.
Med. Genet. 57: 489-492, 1995.
Suttie, J. W. Vitamin K-dependent carboxylase. Ann. Rev.
Biochem. 54: 459-477, 1985.
Tasker, W. G., Mastri, A. R., Gold, A. P. Chondrodystrophia
calcificans congenita (dysplasia epiphysialis punctata): recognition of the
clinical picture. Am. J. Dis. Child. 119: 122-127, 1970.
Teigler, A., Komljenovic, D., Draguhn, A., Gorgas, K., Just,
W. W. Defects in myelination, paranode organization and Purkinje cell
innervation in the ether lipid-deficient mouse cerebellum. Hum. Molec. Genet.
18: 1897-1908, 2009.
Thai, T.-P., Rodemer, C., Jauch, A., Hunziker, A., Moser,
A., Gorgas, K., Just, W. W. Impaired membrane traffic in defective ether lipid
biosynthesis. Hum. Molec. Genet. 10: 127-136, 2001.
Toriello, H. V. Chondrodysplasia punctata and maternal
systemic lupus erythematosus. (Commentary) J. Med. Genet. 35: 698-699, 1998.
Toriello, H. V., Higgins, J. V., Miller, T. Provisionally
unique autosomal recessive chondrodysplasia punctata syndrome. Am. J. Med.
Genet. 47: 797-799, 1993.
Traupe, H. Functional X-chromosomal mosaicism of the skin:
Rudolf Happle and the lines of Alfred Blaschko. Am. J. Med. Genet. 85: 324-329,
1999.
Traupe, H., Muller, D., Atherton, D., Kalter, D. C.,
Cremers, F. P. M., van Oost, B. A., Ropers, H.-H. Exclusion mapping of the
X-linked dominant chondrodysplasia punctata/ichthyosis/cataract/short stature
(Happle) syndrome: possible involvement of an unstable pre-mutation. Hum.
Genet. 89: 659-665, 1992
Trowitzsch, E., Richter, R., Eisenberg, W., Kallfelz, H. C.
Severe pulmonary arterial stenoses in Conradi-Hunermann disease. Europ. J.
Pediat. 145: 116-118, 1986
Uwechue, I. C., Cooper, B. F., Goble, C., Hacker, T., Blair,
H. J., Burke, D. T., Herman, G., Boyd, Y. The mouse X-linked developmental
mutant, tattered, lies between DXMit55 and Xkh and is associated with
hyperkeratinization. Genomics 37: 238-241, 1996.
Van Maldergem, L., Espeel, M., Roels, F., Petit, C.,
Dacremont, G., Wanders, R. J. A., Verloes, A., Gillerot, Y. X-linked recessive
chondrodysplasia punctata with XY translocation in a stillborn fetus. Hum.
Genet. 87: 661-664, 1991.
Vinke, T. H., Duffy, F. P. Chondrodystrophia calcificans
congenita: report of 2 cases. J. Bone Joint Surg. Am. 29: 509-514, 1974.
Viseskul, C., Opitz, J. M., Spranger, J. W., Hartmann, H.
A., Gilbert, E. F. Pathology of chondrodysplasia punctata rhizomelic type.In:
Bergsma, D. : Skeletal Dysplasias. Amsterdam: Excerpta Medica (pub.) 1974. Pp.
327-333.
Wanders, R. J. A., Dekker, C., Hovarth, V. A. P., Schutgens,
R. B. H., Tager, J. M., Van Laer, P., Lecoutere, D. Human
alkyldihydroxyacetonephosphate synthase deficiency: a new peroxisomal disorder.
J. Inherit. Metab. Dis. 17: 315-318, 1994
Wanders, R. J. A., Saelman, D., Heymans, H. S. A.,
Schutgens, R. B. H., Westerveld, A., Poll-The, B. T., Saudubray, J. M., Van den
Bosch, H., Strijland, A., Schram, A. W., Tager, J. M. Genetic relation between
the Zellweger syndrome, infantile Refsum's disease, and rhizomelic
chondrodysplasia punctata. (Letter) New Eng. J. Med. 314: 787-788, 1986
Wanders, R. J. A., Schumacher, H., Heikoop, J., Schutgens,
R. B. H., Tager, J. M. Human dihydroxyacetonephosphate acyltransferase
deficiency: a new peroxisomal disorder. J. Inherit. Metab. Dis. 15: 389-391,
1992
Wanders, R. J. A., Waterham, H. R. Peroxisomal disorders I:
biochemistry and genetics of peroxisome biogenesis disorders. Clin. Genet. 67:
107-133, 2005.
Wardinsky, T. D., Pagon, R. A., Powell, B. R., McGillivray,
B., Stephan, M., Zonana, J., Moser, A. Rhizomelic chondrodysplasia punctata and
survival beyond one year: a review of the literature and five case reports.
Clin. Genet. 38: 84-93, 1990.
Waterham, H. R., Ebberink, M. S. Genetics and molecular
basis of human peroxisome biogenesis disorders. Biochim. Biophys. Acta 1822:
1430-1441, 2012.
Weil, D., Portnoi, M.-F., Levilliers, J., Wang, I., Mathieu,
M., Taillemite, J.-L., Meier, M., Boudailliez, B., Petit, C. A 45,X male with
an X;Y translocation: implications for the mapping of the genes responsible for
Turner syndrome and X-linked chondrodysplasia punctata. Hum. Molec. Genet. 2:
1853-1856, 1993.
Wettke-Schafer, R., Kantner, G. X-linked dominant inherited
diseases with lethality in hemizygous males. Hum. Genet. 64: 1-23, 1983.
White, A. L., Modaff, P., Holland-Morris, F., Pauli, R. M.
Natural history of rhizomelic chondrodysplasia punctata. Am. J. Med. Genet.
118A: 332-342, 2003
Whitfield, M. F. Chondrodysplasia punctata after warfarin in
early pregnancy: case report and summary of the literature. Arch. Dis. Child.
55: 139-142, 1980.
Wulfsberg, E. A., Curtis, J., Jayne, C. H. Chondrodysplasia
punctata: a boy with X-linked recessive chondrodysplasia punctata due to an
inherited X-Y translocation with a current classification of these disorders.
Am. J. Med. Genet. 43: 823-828, 1992
_ga1tj5as.jpg)
Aggiornamenti
- Patologie Genetiche dello Scheletro
Sono elencate tutte le 436 Displasie Scheletriche con la... - Consigli per l'utilizzo del software diagnosi in Diagnosi...
Consigli per l'utilizzo del software diagnosi in Diagnosi... - TERMINI DI USO DEL PORTALE WEB med2000eco e Software...
TERMINI DI USO DEL PORTALE WEB med2000eco e Software...