![]()
Malattia di Gaucher tipo II OMIN 230900
Malattia di Gaucher tipo II OMIM 230900
E' una rara malattia genetica dovuta a deficit del gene che codifica per la beta-glucosidasi acida. E distinta in tre tipi: nel Tipo 1 il deficit dell'enzima beta-glucosidasi provoca accumulo di glucocerebroside nelle cellule reticoloendoteliali di fegato, milza e midollo; nei Tipi 2 e 3 il deficit dell'enzima è causa dell'interessamento anche del Sistema Nervoso Centrale con manifestazioni neurologiche che nel Tipo 2 provocano una grave compromissione del SNC con exitus precoce.
La Diagnosi Prenatale Ecografica si basa su:
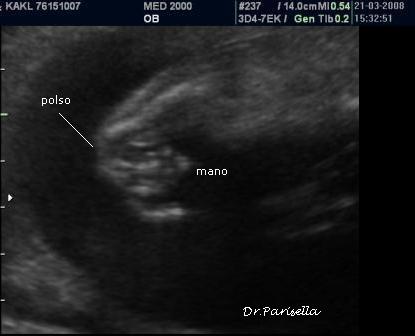
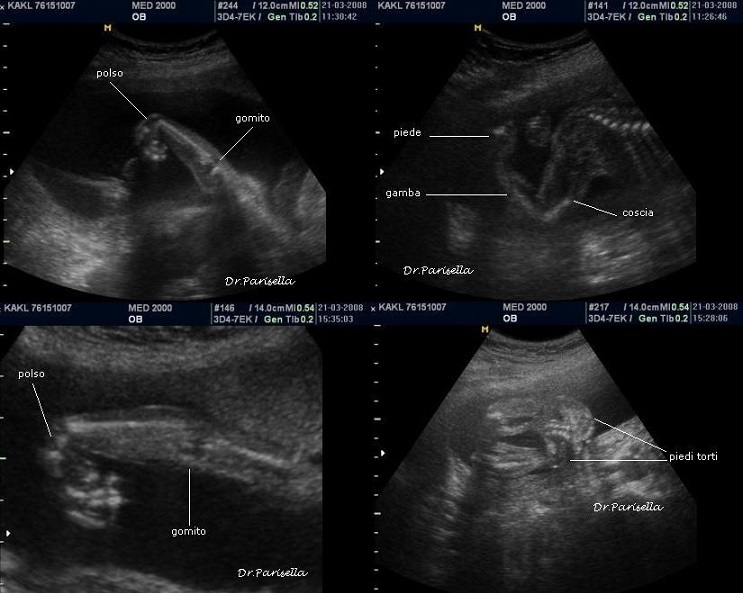
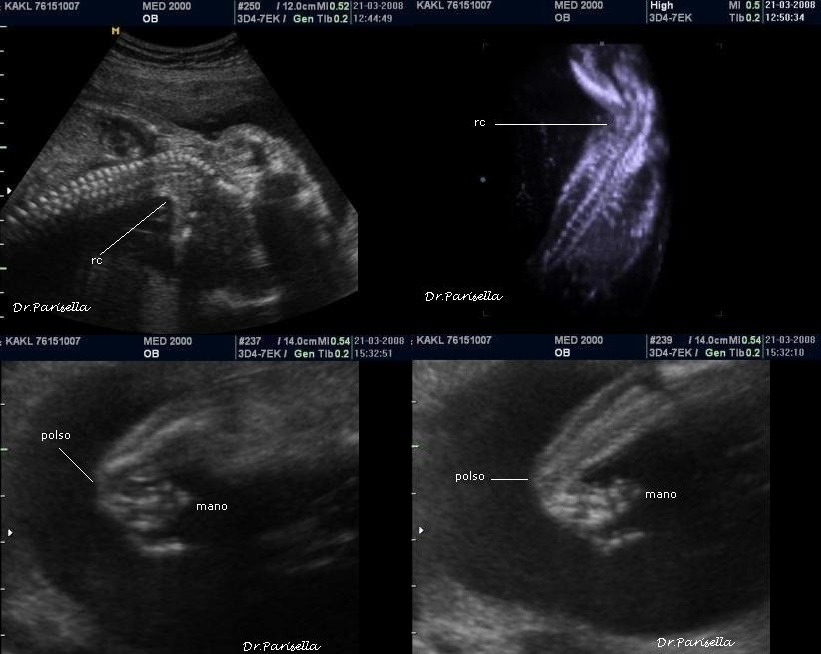
1) assenza di movimenti attivi fetali.
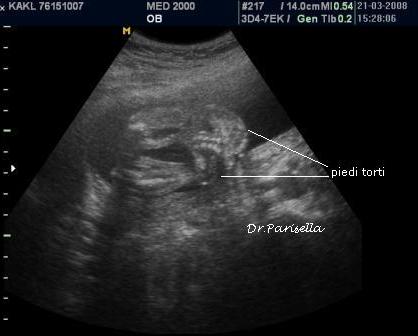
2) contratture multiple.
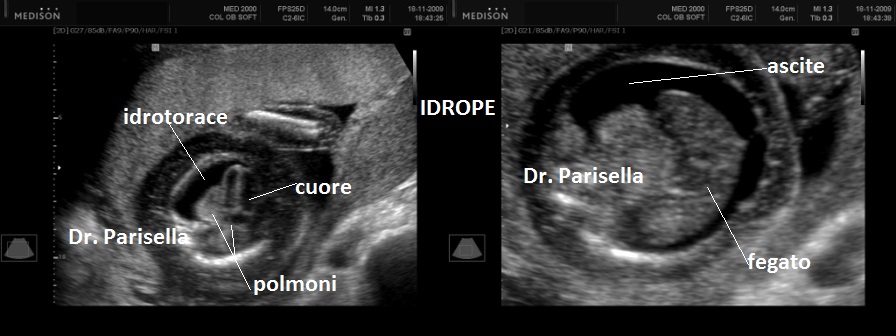
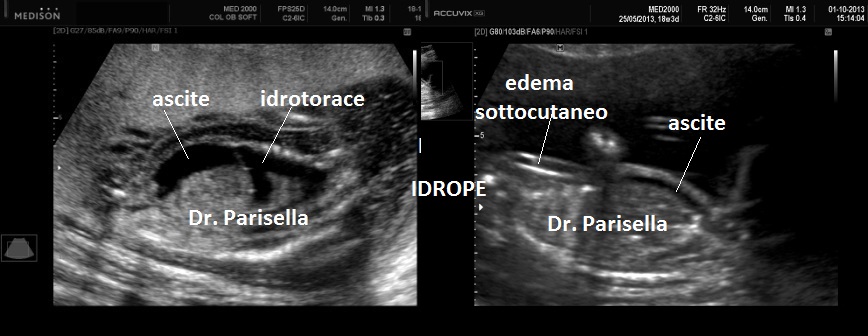
3) Idrope.
E possibile la diagnosi
enzimatica sul liquido amniotico oppure lo studio molecolare delle mutazioni
mediante prelievo di villi coriali.
Bibliografia
Beutler, E., Kuhl, W.Glucocerebrosidase
processing in normal fibroblasts and in fibroblasts from patients with type I,
type II, and type III Gaucher disease. Proc. Nat. Acad. Sci. 83: 7472-7474,
1986.
Owada, M., Sakiyama,
T., Kitagawa, T. Neuropathic Gaucher's disease with normal
4-methylumbelliferyl-beta-glucosidase activity in the liver. J. Pediat. Res.
11: 641-646, 1977.
Schneider, E. L.,
Ellis, W. G., Brady, R. O., McCulloch, J. R., Epstein, C. J. Infantile (type
II) Gaucher's disease: in utero diagnosis and fetal pathology. J. Pediat. 81:
1134-1139, 1972.
Stone, D. L., Tayebi,
N., Orvisky, E., Stubblefield, B., Madike, V., Sidransky, E. Glucocerebrosidase
gene mutations in patients with type 2 Gaucher disease. Hum. Mutat. 15:
181-188, 2000.
Aggiornamenti
- Patologie Genetiche dello Scheletro
Sono elencate tutte le 436 Displasie Scheletriche con la... - Consigli per l'utilizzo del software diagnosi in Diagnosi...
Consigli per l'utilizzo del software diagnosi in Diagnosi... - TERMINI DI USO DEL PORTALE WEB med2000eco e Software...
TERMINI DI USO DEL PORTALE WEB med2000eco e Software...