![]()
Osteogenesi Imperfetta
Osteogenesi imperfetta
redatta da Dr. P.Parisella
L'osteogenesi imperfetta è una malattia
rara (incidenza 1 su 10.000/20.000) conosciuta come "la
malattia delle ossa fragili" perché
caratterizzata da una riduzione della resistenza delle ossa che le rende più
suscettibili alle fratture. Nella maggior parte dei casi la riduzione di
resistenza delle ossa è legata ad alterazioni del tessuto connettivo
conseguenti a mutazioni dei geni COL1A1 e COL1A2 che contengono le informazioni
per sintetizzare il collagene.
Genetica
La mutazione genetica può avvenire de novo o trasmessa con modalità
autosomica dominante (genitore affetto ai figli con una probabilità del 50% ad
ogni gravidanza).
Sulla base delle caratteristiche clinico/radiologiche
erano stati definiti 4 tipi di osteogenesi imperfetta tutti determinati da
mutazioni nei geni COL1A1 e COL1A2.
- Tipo
1: forma non deformante con sclere blu.
Forma lieve, con fratture che iniziano con la deambulazione e si riducono dopo
la pubertà. La statura è quasi normale. E' presente diminuzione della
percezione uditiva (ipoacusia) in età avanzata;
- Tipo 2: forma congenita o
letale perinatale. I neonati presentano ossa corte e incurvate per le
numerose fratture che si sono accumulate in epoca prenatale. Sono presenti
sclere blu e dentinogenesi imperfetta. Causa di morte è l'insufficienza
respiratoria;
- Tipo 3: forma progressivamente
deformante. Forma grave, ma non letale. Sono presenti
dentinogenesi imperfetta, compressione vertebrale e scoliosi, bassa statura;
- Tipo 4: forma comune moderata
con sclere normali. Presenta gravità lieve-moderata (più grave del tipo
1, ma meno grave dei tipi 2 e 3), bassa statura, dentinogenesi imperfetta.
Nel corso degli anni sono state identificate mutazioni di altri geni. E' stato
aggiunto il
tipo 5 legato a mutazioni del gene IFIMT5 caratterizzata
da bassa statura, dislocazione della testa del radio, sclere bianche e
alterazioni istologiche caratteristiche; l'Osteogenesi imperfetta di tipo 5
viene trasmessa con modalità autosomica-dominante.
I tipi di osteogenesi identificati più di recente (tipi da 6 a 18) sono stati definiti in base al gene alterato ed hanno manifestazioni cliniche simili ai tipi 2, 3, 4:
Forme
con manifestazioni simili al tipo 2: tipo 7, tipo 8 e tipo 9;
Forme con manifestazioni simili al tipo 3: tipo 6,
tipo 7, tipo 8, tipo 9, tipo 10, tipo 11, tipo 12, tipo 14, tipo 15 e tipo 16);
Forme con manifestazioni simili al tipo 4: tipo 7,
tipo 9, tipo 13 e tipo 15.
Lista dei geni interessati
OSTEOGENESI IMPERFETTA TIPO I COL1A1 OSTEOGENESI IMPERFETTA TIPO II COL1A1 OSTEOGENESI IMPERFETTA TIPO II COL1A2 OSTEOGENESI IMPERFETTA TIPO IIA COL1A1 OSTEOGENESI IMPERFETTA TIPO IIB CRTAP OSTEOGENESI IMPERFETTA TIPO IIC COL1A1OSTEOGENESI IMPERFETTA TIPO III COL1A1 OSTEOGENESI IMPERFETTA TIPO III COL1A2 OSTEOGENESI IMPERFETTA TIPO III/IV COL1A1 OSTEOGENESI IMPERFETTA TIPO IV COL1A1 OSTEOGENESI IMPERFETTA TIPO IV COL1A2 OSTEOGENESI IMPERFETTA TIPO 5 IFIMT5OSTEOGENESI IMPERFETTA TIPO 6 SERPINF1OSTEOGENESI IMPERFETTA TIPO 7 CRTAP OSTEOGENESI IMPERFETTA TIPO 8 P3H1 OSTEOGENESI IMPERFETTA TIPO 9 PPIBOSTEOGENESI IMPERFETTA TIPO 10 SERPINH1OSTEOGENESI IMPERFETTA TIPO 11 FKBP10OSTEOGENESI IMPERFETTA TIPO 12 BMP1OSTEOGENESI IMPERFETTA TIPO 13 SP7OSTEOGENESI IMPERFETTA TIPO 14 TMEM38BOSTEOGENESI IMPERFETTA TIPO 15 WNT1OSTEOGENESI IMPERFETTA TIPO 16 CREB3LI
Diagnosi Ecografica
Quella che interessa dal
punto di vista della diagnosi ecografica prenatale è l'Osteogenesi Imperfetta
tipo II (OMIM 166210) caratterizzata da fragilità ossea con fratture multiple,
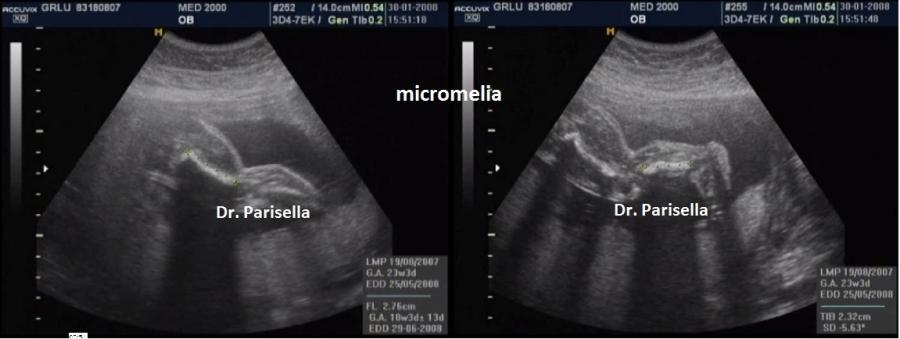
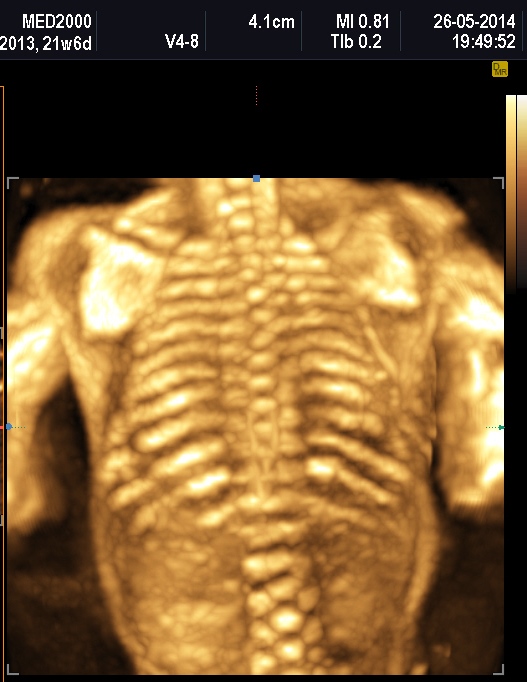
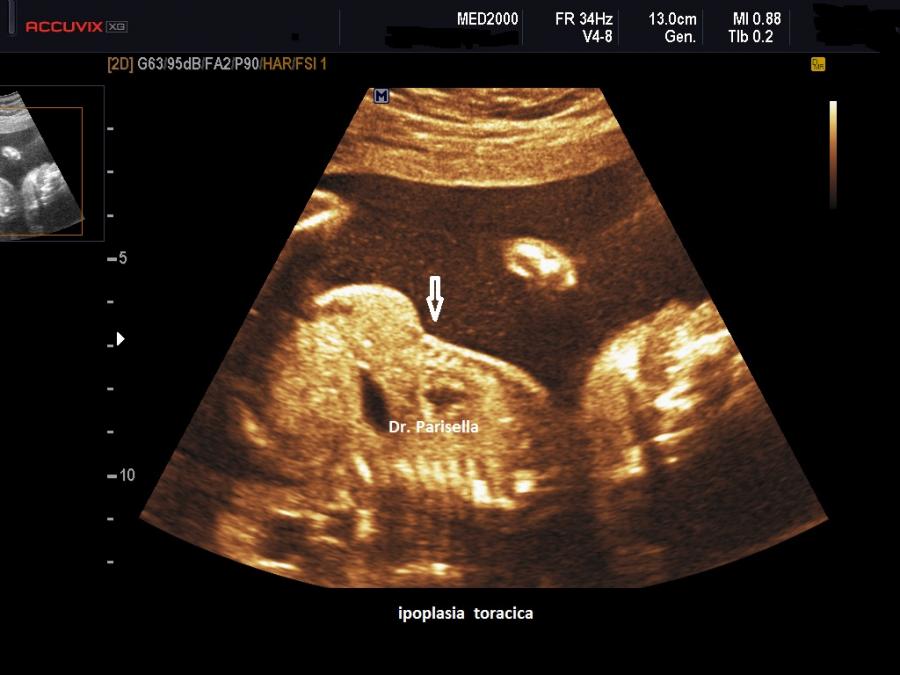
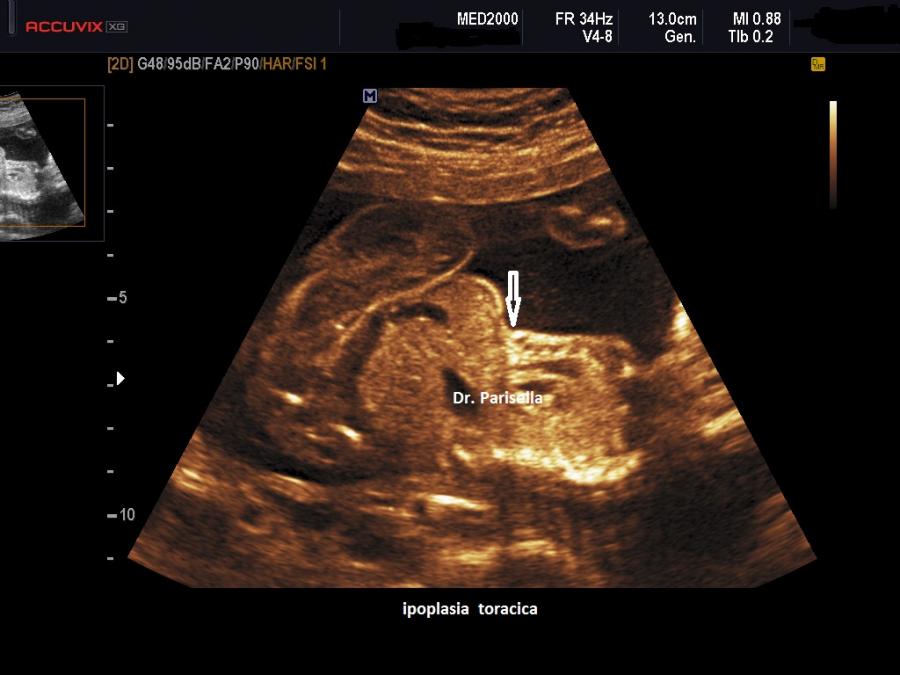
micromelia severa dovuta alle fratture delle ossa lunghe, ipoplasia toracica
severa a volte con fratture costali, ipomineralizzazione diffusa del
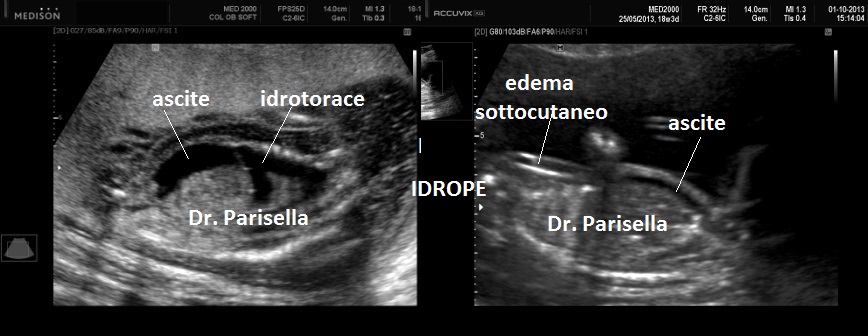
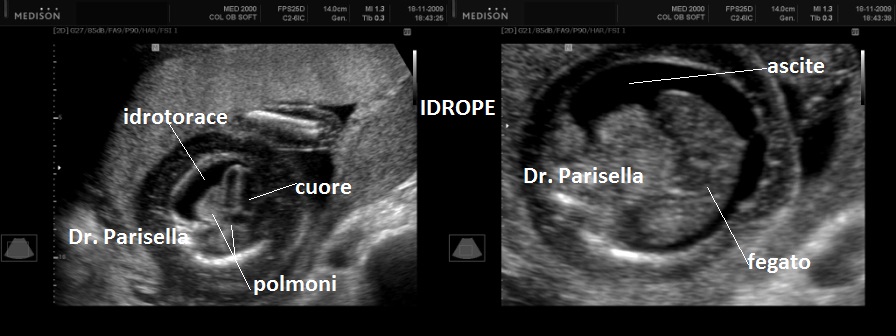
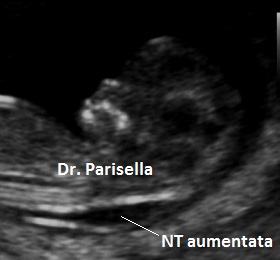
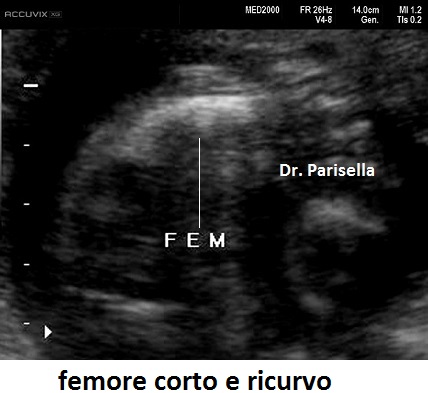
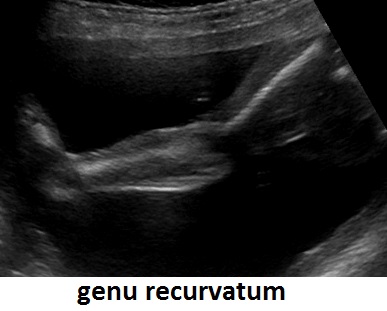
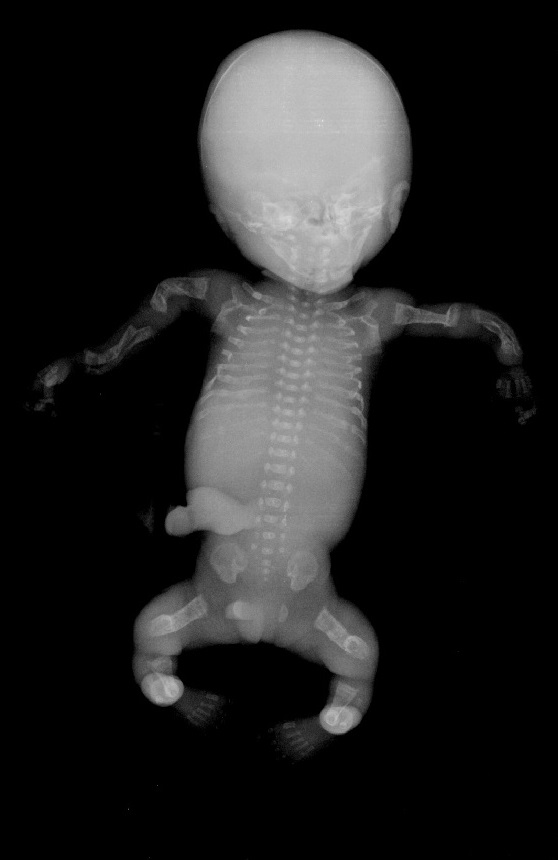
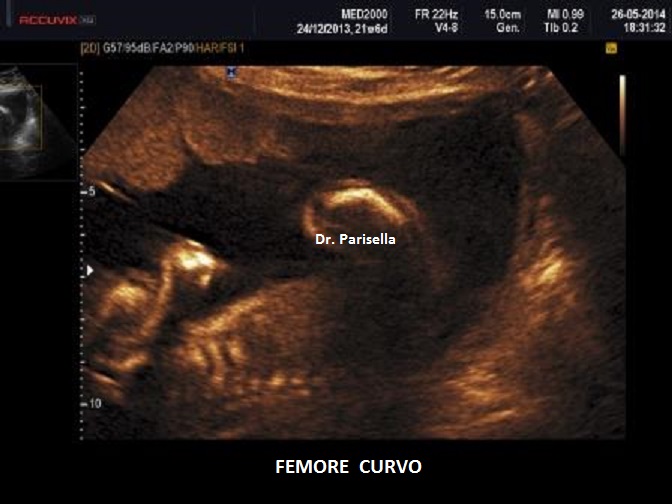
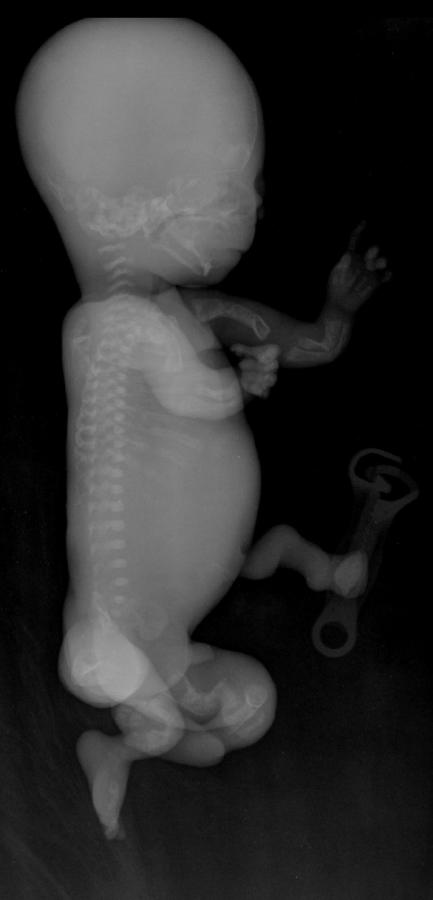
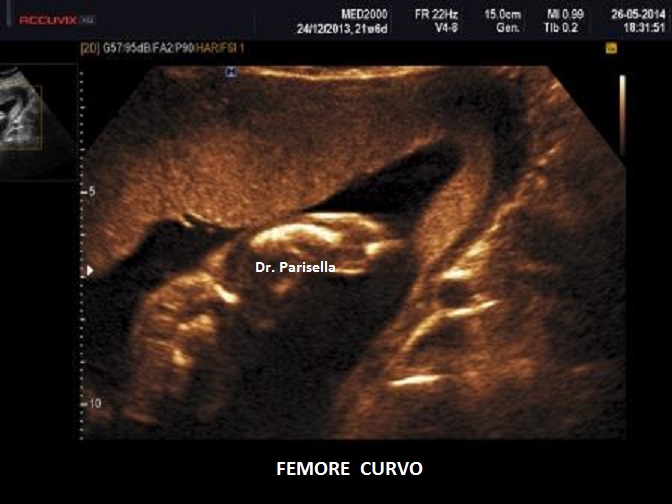
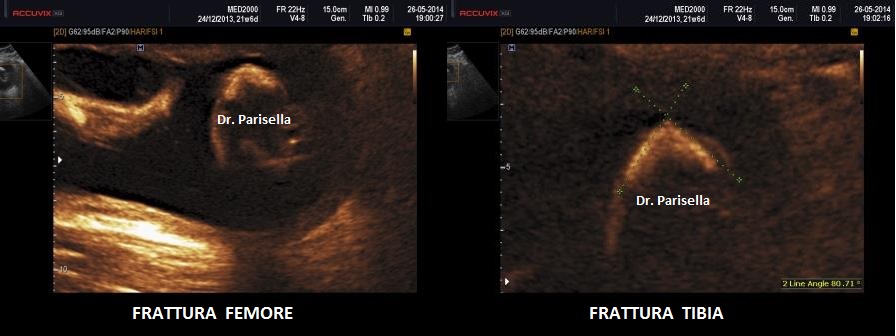
cranio. Ecograficamente si manifesta con ossa corte, ricurve e
fratturate (le ossa fratturate si presentano angolate); ossa craniche
scarsamente ossificate (tanto che la volta cranica può avere una ecogenicità
simile a quella della linea mediana) con conseguente migliore evidenza e
definizione delle strutture cerebrali e segno caratteristico la deformabilità
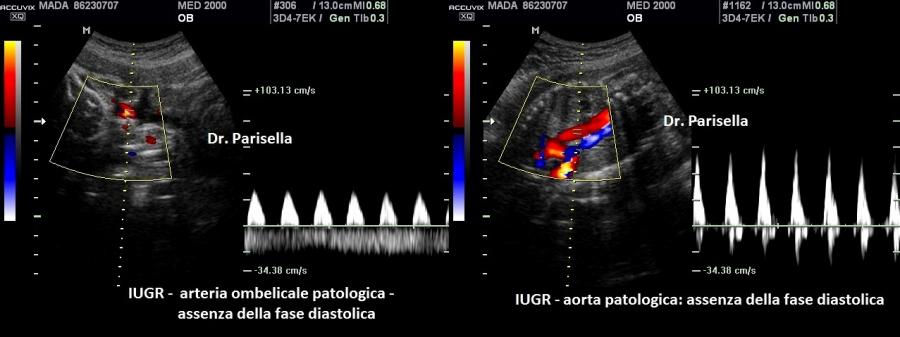
della teca cranica.; ipoplasia toracica con fratture costali; IUGR; movimenti
fetali scarsi.
Diagnosi
Differenziale
Quella che interessa dal
punto di vista della diagnosi ecografica prenatale è l'Osteogenesi Imperfetta
tipo II (OMIM 166210) caratterizzata da fragilità ossea con fratture multiple,
micromelia severa dovuta alle fratture delle ossa lunghe, ipoplasia toracica
severa a volte con fratture costali, ipomineralizzazione diffusa del
cranio. Ecograficamente si manifesta con ossa corte, ricurve e
fratturate (le ossa fratturate si presentano angolate); ossa craniche
scarsamente ossificate (tanto che la volta cranica può avere una ecogenicità
simile a quella della linea mediana) con conseguente migliore evidenza e
definizione delle strutture cerebrali e segno caratteristico la deformabilità
della teca cranica.; ipoplasia toracica con fratture costali; IUGR; movimenti
fetali scarsi.
1) Acondrogenesi tipo II dove non sono
presenti le fratture. 2) Ipofosfatasia dove la ipomineralizzazione è massima mentre le fratture sono
molto più frequenti nella Osteogenesi Imperfetta.3) Displasia Tanatofora tipo I dove non sono presenti le fratture.
Prognosiletale con exitus per insufficienza respiratoria
Byers, P. H., Krakow,
D., Nunes, M. E., Pepin, M. Genetic evaluation of suspected osteogenesis
imperfecta (OI). Genet. Med. 8: 383-388, 2006.
Cohn, D. H., Starman,
B. J., Blumberg, B., Byers, P. H. Recurrence of lethal osteogenesis imperfecta
due to parental mosaicism for a dominant mutation in a human type I collagen
gene (COL1A1). Am. J. Hum. Genet. 46: 591-601, 1990.
Cole, W. G.,
Dalgleish, R. Perinatal lethal osteogenesis imperfecta. J. Med. Genet. 32:
284-289, 1995.
Daw, S. C. M., Gibbs,
D. A., Nicholls, A. C., Hall, E. C., Siggers, D. C., Pope, F. M. Lethal
osteogenesis imperfecta: a family with 6 affected sibs heterozygous for a type
I collagen mutation. (Abstract) J. Med. Genet. 27: 206, 1990.
Elejalde, B. R.,
Mercedes de Elejalde, M. Prenatal diagnosis of perinatally lethal osteogenesis
imperfecta. Am. J. Med. Genet. 14: 353-359, 1983.
Heller, R. H., Winn,
K. J., Heller, R. M. The prenatal diagnosis of osteogenesis imperfecta
congenita. Am. J. Obstet. Gynec. 121: 572-573, 1975.
Horan, F., Beighton,
P. Autosomal recessive inheritance of osteogenesis imperfecta. Clin. Genet. 8:
107-111, 1975.
Shapiro, J. E.,
Phillips, J. A., Byers, P. H., Sanders, R., Holbrook, K. A., Levin, L. S.,
Dorst, J., Barsh, G. S., Peterson, K. E., Goldstein, P. Prenatal diagnosis of
lethal perinatal osteogenesis imperfecta (OI type II). J. Pediat. 101: 127-133,
1982.
Sillence, D. O.,
Barlow, K. K., Garber, A. P., Hall, J. G., Rimoin, D. L. Osteogenesis
imperfecta type II: delineation of the phenotype with reference to genetic
heterogeneity. Am. J. Med. Genet. 17: 407-423, 1984.
Sillence, D. O.,
Senn, A., Danks, D. M. Genetic heterogeneity in osteogenesis imperfecta. J.
Med. Genet. 16: 101-116, 1979.
Stephens, J. D.,
Filly, R. A., Callen, P. W., Golbus, M. S. Prenatal diagnosis of osteogenesis
imperfecta type II by real-time ultrasound. Hum. Genet. 64: 191-193, 1983.


Aggiornamenti
- Patologie Genetiche dello Scheletro
Sono elencate tutte le 436 Displasie Scheletriche con la... - Consigli per l'utilizzo del software diagnosi in Diagnosi...
Consigli per l'utilizzo del software diagnosi in Diagnosi... - TERMINI DI USO DEL PORTALE WEB med2000eco e Software...
TERMINI DI USO DEL PORTALE WEB med2000eco e Software...