![]()
Craniosinostosi
CRANIOSINOSTOSI
Dr. P.Parisella in aggiornamento 2024
E' un gruppo di patologie scheletriche (circa 100), estremamente eterogenee, caratterizzate dalla chiusura prematura di una o più suture craniche. L'incidenza di tutte le forme di craniostenosi è di 1/2.500 nati vivi. La maggior parte delle forme sindromiche (sindrome di Apert, sindrome di Carpenter, sindrome di Crouzon) è causata da mutazioni nei geni per i recettori dei fattori di crescita dei fibroblasti (FGFR) e l'età paterna appare essere correlata ad una maggiore incidenza di malattia.
Fisiologicamente l'espansione delle ossa della volta cranica avviene per deposizione ossea a livello delle suture. Quando il processo di deposizione ossea si interrompe viene impedita la crescita nella direzione della deposizione (cioè in senso perpendicolare alla sutura interessata) e le suture interessate si saldano prematuramente (sinostosi); di conseguenza si verifica una rapida deposizione compensatoria nelle restanti suture che avviene in direzione parallela alle suture chiuse. Ne derivano deformazioni del cranio il cui risultato finale, cioè la conformazione del cranio, dipende dalle suture coinvolte.
 dal web
dal web
La morfologia del cranio varia quindi a seconda delle suture interessate dalla sinostosi:
- acrocefalia o turricefalia: interessamento delle suture coronale, sagittale e lambdoidea con cranio allungato in senso verticale ed accorciato in senso antero-posteriore.
- brachicefalia: interessamento bilaterale della sutura coronale con cranio accorciato in senso antero-posteriore.
- scafocefalia o dolicocefalia: interessamento della sutura sagittale con cranio allungato in senso antero-posteriore.
- plagiocefalia: interessamento monolaterale della sutura coronale (plagiocefalia anteriore) o lambdoidea (plagiocefalia posteriore); in tali casi il cranio ha una forma asimmetrica per appiattimento monolaterale frontale o occipitale.
- cranio a trifoglio: interessamento precoce delle suture coronale, lambdoidea e metopica per cui il cranio assume un aspetto trilobato; si associa ad idrocefalo.
- trigonocefalia: interessamento della sutura metopica con cranio triangolare a punta orientata frontalmente.
- pachicefalia: si ha precoce saldatura delle suture lambdoidee e conseguente appiattimento posteriore del cranio.
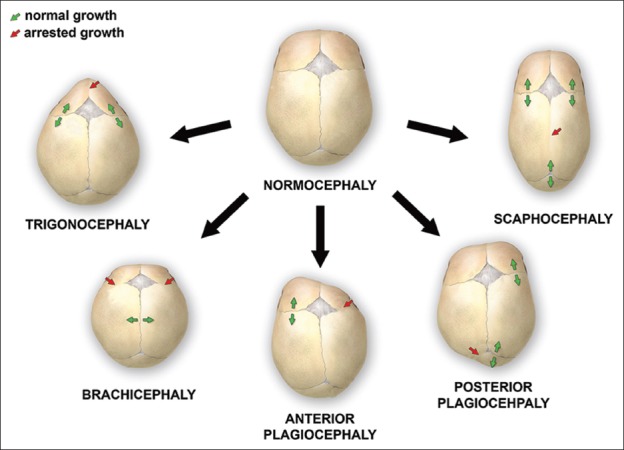 da: Nina Kajdic, et al.Craniosynostosis - Recognition, clinical characteristics, and treatment. Review Bosn J Basic Med Sci . 2018 May 20
da: Nina Kajdic, et al.Craniosynostosis - Recognition, clinical characteristics, and treatment. Review Bosn J Basic Med Sci . 2018 May 20
CLASSIFICAZIONE
Vengono utilizzate diverse classificazioni di craniosinostosi a seconda del meccanismo sottostante, della presenza di altri disturbi o del numero di suture fuse. Ad esempio, se una craniosinostosi si sviluppa a causa di un difetto primario del processo di ossificazione si parla di craniosinostosi primaria. La craniosinostosi secondaria può essere il risultato di malattie sistemiche come il rachitismo e l'ipotiroidismo. La craniosinostosi secondaria può svilupparsi anche nei neonati con microcefalia a causa di un fallimento della crescita cerebrale o in seguito al posizionamento di shunt nei bambini con idrocefalo. Inoltre la craniosinostosi può essere classificata in craniosinostosi sindromica, ad esempio come parte della sindrome di Apert, Crouzon o Pfeiffer, e craniosinostosi non sindromica, dove si sviluppa come disturbo isolato. Craniosinostosi semplice è un termine usato quando solo una sutura si fonde prematuramente, mentre craniosinostosi complessa viene usata per descrivere una fusione prematura di più suture (Kang SG, et al. 2016; Panchal J, et al. 2003; Slater BJ, et al. 2008.)
Schematicamente possiamo dividerle in due gruppi:
La diagnosi prenatale, nei pochi casi in cui è possibile, è spesso tardiva e posta nel terzo trimestre. Una anomalia frequentemente associata è l'ernia diaframmatica che, se presente, può essere il primo segno.
Da tenere presente che anomalie della forma del cranio possono essere presenti in altre patologie non necessariamente caratterizzate da craniosinostosi, ad es.: brachicefalia nella trisomia 21, nella condrodisplasia punctata e nella sindrome di Cornelia De Lange; cranio a trifoglio nella displasia tanatofora tipo II; cranio a limone nei difetti spinali.
Le 4 forme di Craniosinostosi complesse o sindromiche più frequenti sono: Sindrome di Apert, Sindrome di Carpenter, Sindrome di Crouzon, Sindrome di Pfeiffer.
Sindrome di Apert o Acrocefalosindatilia OMIM 101200
Segni principali: acrocefalia o turricefalia; ipertelorismo; frontal bossing; naso insellato; brachi- sindattilia Segni aggiuntivi: prognatismo; anomalie cardiache; anomalie renali; anomalie delle vertebre cervicali; ernia diaframmatica; encefalocele; polidramnios; NT aumentata
Note diagnostiche: La Sindrome di Apert o Acrocefalosindattilia è una craniosinostosi secondaria alla precoce saldatura della sutura coronale. Il cranio presenta una conformazione caratterizzata da aumento del diametro verticale (caudo-craniale) e riduzione del diametro antero-posteriore con occipite piatto (Acrocefalia).
Sindrome di Carpenter OMIM 201000
Segni principali: sindattilia; polidattilia; brachidattilia; anomalie facciali; acrocefalia.
Diagnosi
La sindrome di Carpenter è una craniosinostosi caratterizzata da acrocefalia, anomalie facciali, brachidattilia, sindattilia delle mani e polisindattilia preassiale dei piedi. Può essere presente onfalocele. E' dovuta alla saldatura precoce delle suture coronale, sagittale e lambdoidea (come nella Sindrome di Crouzon) con conseguente acrocefalia (aumento del diametro verticale del cranio e diminuzione del diametro antero-posteriore).
Segni principali: craniosinostosi (acrocefalia - turricefalia); ipoplasia mascellare; esoftalmo bozze frontali prominenti - frontal bossing; ipertelorismo.
Diagnosi
Sono tipiche della sindrome la PROPTOSI OCULARE marcata (sempre presente, dovuta alla presenza di orbite poco profonde; col passare degli anni può portare a riduzione della vista), L'IPOPLASIA MASCELLARE (che provoca un prognatismo mandibolare relativo e un affollamento dei denti anteriori), e la ACROCEFALIA (sinostosi a carico della sutura lamboidea, coronale e sagittale) con conseguente cranio allungato in senso verticale ed accorciato in senso antero-posteriore e laterale.
Si trasmette con carattere autosomico dominante da mutazione del gene FGFR2.
Segni principali:
TIPO I TIPO II TIPO III
brachicefalia cranio a trifoglio brachicefalia
ipertelorismo ipertelorismo ipertelorismo
......... esoftalmo esoftalmo
pollici larghi pollici larghi pollici larghi
alluci grandi alluci grandi alluci grandi
brachidattilia brachidattilia brachidattilia
ipoplasia mascellare .................... ....................
........... sindattilia ...........
Note diagnostiche:
La sindrome di Pfeiffer è stata suddivisa in tre diverse forme: il tipo I più frequente e meno grave, ed i tipi II e III.
E’ legata a mutazioni dei geni FGFR1 e FGFR2
BIBLIOGRAFIA
Governale LS. Craniosynostosis. Pediatr Neurol. 2015;53(5):394–401.
Kang SG, Kang JK. Current and future perspectives in craniosynostosis. J Korean Neurosurg Soc. 2016;59(3):247–9.
Kim HJ, Roh HG, Lee IW. Craniosynostosis: Updates in radiologic diagnosis. J Korean Neurosurg Soc. 2016;59(3):219–26.
Kjaer I. Neuro-osteology. Crit Rev Oral Biol Med. 1998;9(2):224–44.
Kouskoura T, Fragou N, Alexiou M, John N, Sommer L, Graf D, et al. The genetic basis of craniofacial and dental abnormalities. Schweiz Monatsschr Zahnmed. 2011;121(7-8):636–46.
Johnson D, Wilkie AO. Craniosynostosis. Eur J Human Genet. 2011;19(4):369–76.
Marie PJ, Kaabeche K, Guenou H. Roles of FGFR2 and twist in human craniosynostosis: Insights from genetic mutations in cranial osteoblasts. Front Oral Biol. 2008;12:144–59.
Miller C, Losken HW, Towbin R, Bowen A, Mooney MP, Towbin A, et al. Ultrasound diagnosis of craniosynostosis. Cleft Palate Craniofac J. 2002;39(1):73–80.
Nina Kajdic , Peter Spazzapan, Tomaz Velnar. Craniosynostosis - Recognition, clinical characteristics, and treatment. Review Bosn J Basic Med Sci . 2018 May 20;18(2):110-116.
Panchal J, Uttchin V. Management of craniosynostosis. Plast Reconstr Surg. 2003;111(6):2032–48.
Slater BJ, Lenton KA, Kwan MD, Gupta DM, Wan DC, Longaker MT. Cranial sutures: A brief review. Plast Reconstr Surg. 2008;121(4):170e–8.
Stelnicki EJ, Mooney MP, Losken HW, Zoldos J, Burrows AM, Kapucu R, et al. Ultrasonic prenatal diagnosis of coronal suture synostosis. J Craniofac Surg. 1997;8(4):252–8.
Zaleckas L, Neverauskien? A, Daugelavicius V, Šidlovskait?-Baltak? D, Raugalas R, Vištartait? B, et al. Diagnosis and treatment of craniosynostosis: Vilnius team experience. Acta Med Litu. 2015;22(2):111–21.
Sindrome di Apert
Chang, C.-C., Tsai, F.-J., Tsai, H.-D., Tsai, C.-H., Hsieh, Y.-Y., lee, C.-C., Yang, T.-C., Wu, J.-Y. Prenatal diagnosis of Apert syndrome. Prenatal Diag. 18: 621-625, 1998.
Cohen, M. M., Jr., Kreiborg, S. Visceral anomalies in the Apert syndrome. Am. J. Med. Genet. 45: 758-760, 1993.
Cohen, M. M., Jr., Kreiborg, S. Hands and feet in the Apert syndrome. Am. J. Med. Genet. 57: 82-96, 1995.
Cohen, M. M., Jr., Kreiborg, S. The central nervous system in the Apert syndrome. Am. J. Med. Genet. 35: 36-45, 1990.
Kreiborg, A., Barr, M., Jr., Cohen, M. M., Jr. Cervical spine in the Apert syndrome. Am. J. Med. Genet. 43: 704-708, 1992.
Moloney, D. M., Slaney, S. F., Oldridge, M., Wall, S. A., Sahlin, P., Stenman, G., Wilkie, A. O. M. Exclusive paternal origin of new mutations in Apert syndrome. Nature Genet. 13: 48-53, 1996.
Quintero-Rivera, F., Robson, C. D., Reiss, R. E., Levine, D., Benson, C. B., Mulliken, J. B., Kimonis, V. E. Intracranial anomalies detected by imaging studies in 30 patients with Apert syndrome. (Letter) Am. J. Med. Genet. 140A: 1337-1338, 2006.
von Gernet, S., Golla, A., Ehrenfels, Y., Schuffenhauer, S., Fairley, J. D. Genotype-phenotype analysis in Apert syndrome suggests opposite effects of the two recurrent mutations on syndactyly and outcome of craniofacial surgery. Clin. Genet. 57: 137-139, 2000.
Sindrome di Carpenter
Altunhan, H., Annagur, A., Ors, R. The association of Carpenter syndrome and situs inversus totalis: first case report. Turk. Klin. J. Med. Sci. 31: 464-467, 2011.
Cohen, D. M., Green, J. G., Miller, J., Gorlin, R. J., Reed, J. A. Acrocephalopolysyndactyly type II--Carpenter syndrome: clinical spectrum and an attempt at unification with Goodman and Summitt syndromes. Am. J. Med. Genet. 28: 311-324, 1987.
Eaton, A. P., Sommer, A., Kontras, S. B., Sayers, M. P. Carpenter syndrome--acrocephalopolysyndactyly type II. Birth Defects Orig. Art. Ser. 10: 249-260, 1974.
Palacios, E., Schimke, R. N. Craniosynostosis--syndactylism. Am. J. Roentgen. 106: 144-155, 1969.
Robinson, L. K., James, H. E., Mubarak, S. J., Allen, E. J., Jones, K. L. Carpenter syndrome: natural history and clinical spectrum. Am. J. Med. Genet. 20: 461-469, 1985.
Temtamy, S. A. Carpenter's syndrome: acrocephalopolysyndactyly. An autosomal recessive syndrome. J. Pediat. 69: 111-120, 1966.
Sindrome di Crouzon
Cohen, M. M., Jr., Kreiborg, S. Birth prevalence studies of the Crouzon syndrome: comparison of direct and indirect methods. Clin. Genet. 41: 12-15, 1992.
Cohen, S. R., Dauser, R. C., Gorski, J. L. Insidious onset of familial craniosynostosis. Cleft-Palate Craniofac. J. 30: 401-405, 1993.
Kreiborg, S., Cohen, M. M., Jr. Germinal mosaicism in Crouzon syndrome. Hum. Genet. 84: 487-488, 1990.
Kreiborg, S., Jensen, B. L. Variable expressivity of Crouzon's syndrome within a family. Scand. J. Dent. Res. 85: 175-184, 1977.
Meyers, G. A., Orlow, S. J., Munro, I. R., Przylepa, K. A., Jabs, E. W. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nature Genet. 11: 462-464, 1995.
Sindrome di Pfeiffer
Baraitser, M., Bowen-Bravery, M., Saldana-Garcia, P. Pitfalls of genetic counselling in Pfeiffer's syndrome. J. Med. Genet. 17: 250-256, 1980.
Cohen, M. M., Jr. Pfeiffer syndrome update, clinical subtypes, and guidelines for differential diagnosis. Am. J. Med. Genet. 45: 300-307, 1993.
Rutland, P., Pulleyn, L. J., Reardon, W., Baraitser, M., Hayward, R., Jones, B., Malcolm, S., Winter, R. M., Oldridge, M., Slaney, S. F., Poole, M. D., Wilkie, A. O. M. Identical mutations in the FGFR2 gene cause both Pfeiffer and Crouzon syndrome phenotypes. Nature Genet. 9: 173-176, 1995.
Saldino, R. M., Steinbach, H. L., Epstein, C. J. Familial acrocephalosyndactyly (Pfeiffer syndrome). Am. J. Roentgen. 116: 609-622, 1972.
Soekarman, D., Fryns, J. P., van den Berghe, H. Pfeiffer acrocephalosyndactyly syndrome in mother and son with cloverleaf skull anomaly in the child. Genetic Counseling 3: 217-220, 1992.
Stone, P., Trevenen, C. L., Mitchell, I., Rudd, N. Congenital tracheal stenosis in Pfeiffer syndrome. Clin. Genet. 38: 145-148, 1990.
Vallino-Napoli, L. D. Audiologic and otologic characteristics of Pfeiffer syndrome. Cleft Palate Craniofac. J. 33: 524-529, 1996.
Vanek, J., Losan, F. Pfeiffer's type of acrocephalosyndactyly in two families. J. Med. Genet. 19: 289-292, 1982.













Aggiornamenti
- Patologie Genetiche dello Scheletro
Sono elencate tutte le 436 Displasie Scheletriche con la... - Consigli per l'utilizzo del software diagnosi in Diagnosi...
Consigli per l'utilizzo del software diagnosi in Diagnosi... - TERMINI DI USO DEL PORTALE WEB med2000eco e Software...
TERMINI DI USO DEL PORTALE WEB med2000eco e Software...