![]()
Malformazione Adenomatoide Cistica del Polmone (MACP - CCAM - CAM) Sequestro Polmonare
Malformazione Adenomatoide Cistica del Polmone (MACP - CCAM - CAM) - Sequestro Polmonare
Malformazione Adenomatoide Cistica del Polmone (MACP – CCAM – CAM)
La Malformazione Adenomatoide Cistica del Polmone è una rara patologia polmonare caratterizzata dalla presenza di una lesione multicistica o solida, più frequentemente monolaterale, che interessa generalmente un lobo o segmento del polmone.
Se ne distinguono tre tipi (Stocker JT, et al., 1977):
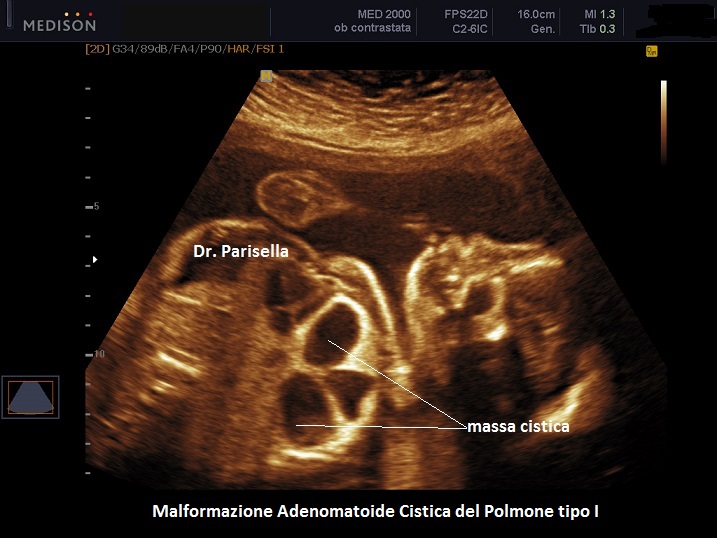
MACP tipo I: è caratterizzata da cisti singole o multiple di dimensioni solitamente > 2,0 cm;
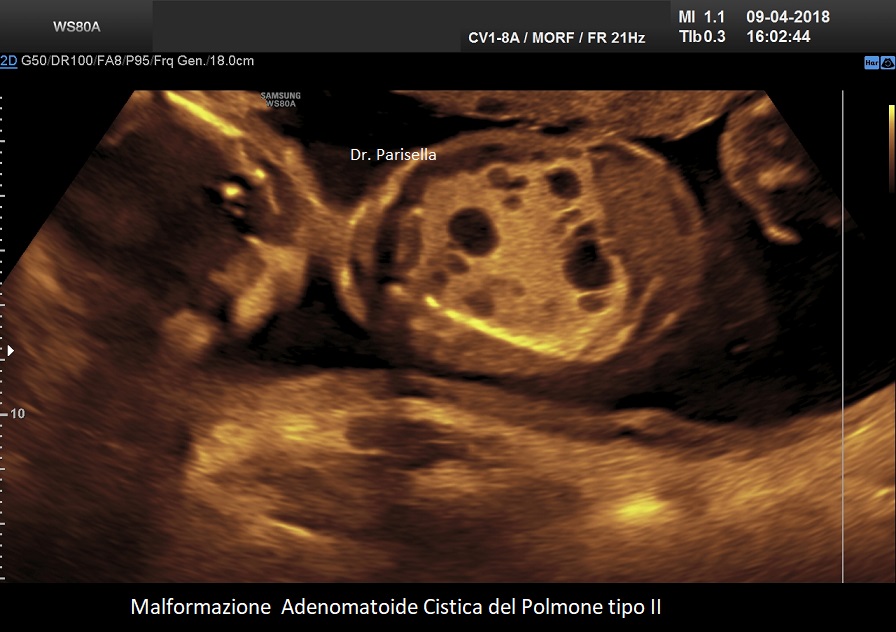
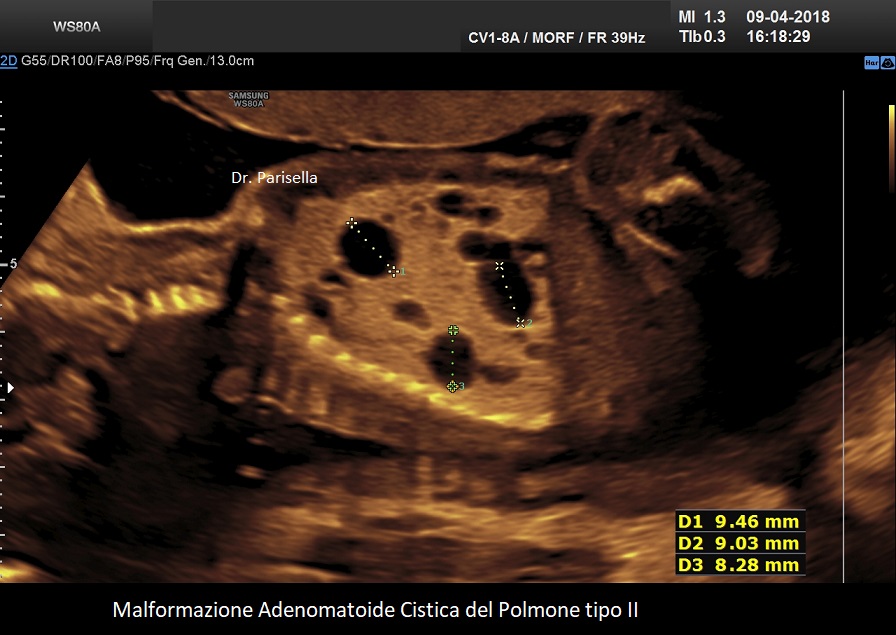
MACP tipo II: è caratterizzato da numerose piccole cisti di diametro < 2,0 cm.;
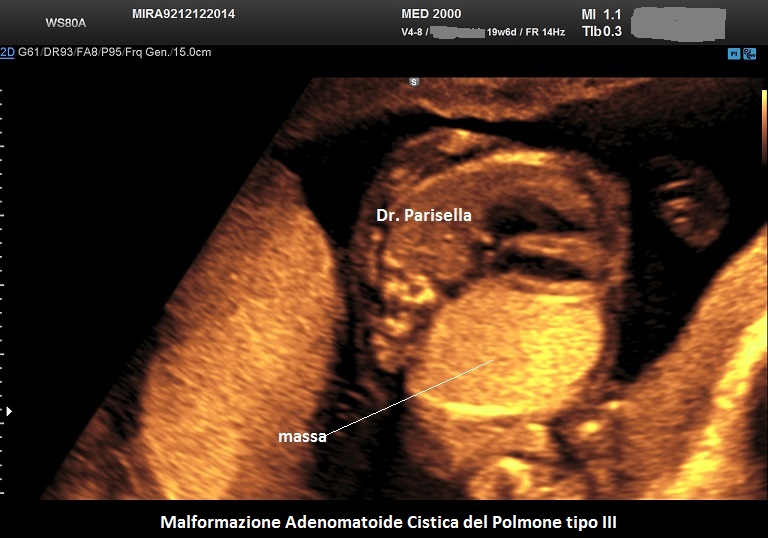
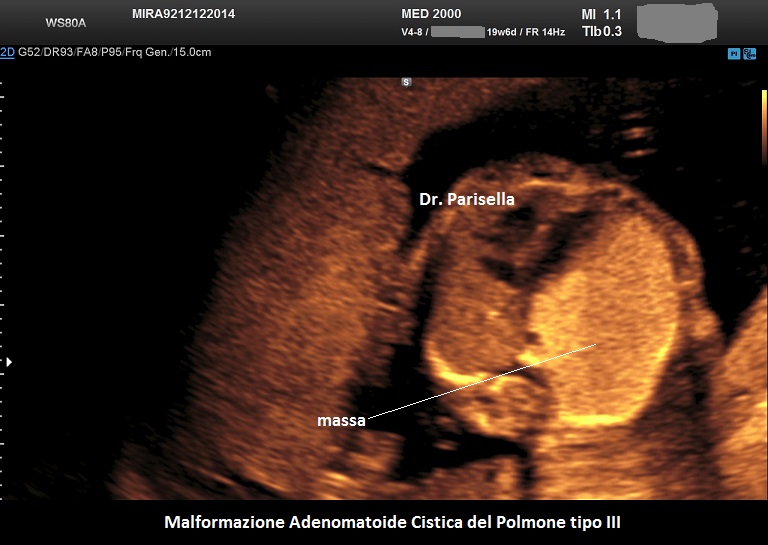
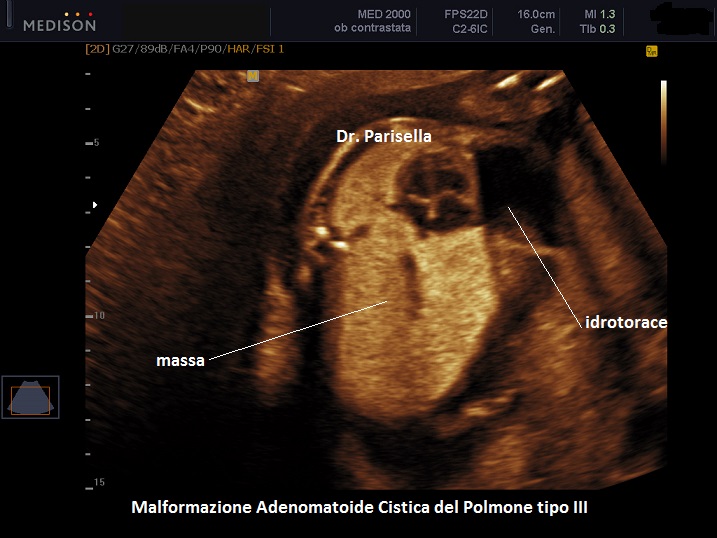
MACP tipo III: è una massa solida che contiene cisti molto piccole < 0,5 cm.
. Il tipo I ha una buona prognosi, è presente in circa il 50% dei casi e le cisti sembrano comunicare con il normale parenchima polmonare e possono essere circondate da cisti più piccole.
. Le lesioni di tipo II si verificano in circa il 40% dei casi e sono costituite da numerose piccole cisti (< 2,0 cm) mescolate con aree di aumentata ecogenicità all'ecografia. La prognosi dipende dalla gravità della lesione e dalla sua associazione con altre malformazioni fetali. Esiste una associazione tra anomalie congenite e CCAM di tipo II, le principali delle quali sono genitourinarie, come la disgenesia o l'agenesia renale, e cardiache, come la tetralogia di Fallot e il tronco arterioso; altri includono l'ernia diaframmatica e le anomalie muscoloscheletriche. Un importante studio, considerato una delle più grandi coorti di casi, conclude che le CCAM microcistiche e macrocistiche presentano entrambe decorsi e prognosi perinatali simili (Chen Y et al., 2021).
. Il tipo III è una massa solida che contiene cisti molto piccole (< 5,0 mm). (Rolo LC et al., 2022).
Fisiopatologia
La CCAM è una malformazione originata da uno sviluppo anomalo delle vie aeree in cui i bronchioli terminali proliferano in modo anomalo all'inizio dello sviluppo embrionale generando cisti intraparenchimali di varie dimensioni (Rolo LC et al., 1992; Leblanc C. et al., 2017; Shamas AG et al., 2017).
Diagnosi ecografica
Si presenta come una massa occupante spazio di solito unilaterale in più del 95% dei casi e solitamente coinvolge un lobo o segmento del polmone; nella sua evoluzione determina dislocazione del mediastino e del cuore. L'ecostruttura distingue due forme: una forma multicistica caratterizzata dalla presenza di multiple cisti di varia grandezza (tipo I e II) e una forma microcistica (tipo III) corrispondente alla variante solida.
La diagnosi differenziale deve essere posta con il Sequestro polmonare: in questo caso è dirimente il riscontro di un vaso afferente originato dall'aorta nel sequestro polmonare a differenza della MACP in cui la massa è irrorata da diramazioni dell'arteria polmonare.
Storia Naturale
La malformazione adenomatoide cistica del polmone (CAM) ha una storia naturale variabile che può evolvere in idrope o regredire. Non sono disponibili criteri per determinare quali lesioni crescerebbero e svilupperebbero idrope rispetto a quelle la cui crescita si stabilizzerebbe o regredirebbe. Per calcolare il rischio di sviluppare l’idrope fetale è stato proposto l’utilizzo del rapporto volumetrico CAM Volume Ratio (CVR) cioè il rapporto tra volume della MACP e la circonferenza cranica (Crombleholme TM et al., 2002). Il volume della MACP viene calcolato moltiplicando i tre diametri della massa x 0,52. I feti con CVR inferiore o uguale a 1,6 sono considerati a basso rischio di sviluppo di idrope e quelli con CVR maggiore di 1,6 sono considerati a rischio aumentato per lo sviluppo dell'idrope. Per valori di CVR > 1,6 il rischio di sviluppare l'idrope è del 75%, mentre per valori di CVR < 1,6 il rischio scende al 16%.
La CVR è un utile indicatore ecografico dei feti a rischio di idrope che richiedono un'attenta osservazione ecografica e un possibile intervento fetale (Tran Hong et al., 2008).
Ricorrenza, Prognosi e Terapia
I rischi di aneuploidia e sindromico sono bassi e non vi è nessun aumento del rischio di ricorrenza.
La diagnosi prenatale di CCAM è associata ad una buona prognosi a breve termine (Tran Hong et al., 2008) con tendenza alla regressione spontanea, in genere nel III trimestre (Shamas AG et al., 2017).
L'uso di corticosteroidi per il trattamento della malformazione adenomatoide polmonare congenita ad alto rischio come opzione terapeutica non invasiva è associato al miglioramento o alla regressione dell'idrope e alla riduzione della CVR, oltre a una riduzione del tasso di mortalità. E’ stato valutato l'effetto dell'uso prenatale di un singolo ciclo di steroidi in pazienti con lesioni prevalentemente microcistiche e/o un CVR >1,6 ed è stata osservata una riduzione della lesione nel 61% dei casi e l'idrope fetale si è risolta nel 78%. (Curran PF et al., 2010). Gli studi dimostrano che, senza il trattamento dei feti con CPAM ad alto rischio, il rischio di sviluppare idrope, che è un importante predittore di mortalità fetale, aumentava fino al 100% (Crombleholme TM et al., 2002; Leblanc C. et al., 2017). I dati disponibili in letteratura dimostrano che il suo utilizzo ha contribuito in modo significativo al miglioramento della prognosi fetale e perinatale ma nonostante i risultati positivi della terapia con corticosteroidi nel trattamento dei feti affetti da CPAM ad alto rischio, è ancora necessario effettuare ulteriori studi sull’argomento (Macedo I., et al. 2022).
Management
La gravidanza dovrebbe inizialmente essere gestita presso un centro di III livello con ecografie seriali. La valutazione ecografica deve essere eseguita settimanalmente per valutare il volume della CCAM e la CVR può identificare i primi segni di idrope fetale. Se sono presenti cisti dominanti, anche con un CVR <1,6, esiste il rischio di crescita acuta della cisti e sviluppo di idrope fetale (Rolo LC et al., 2022). I risultati ecografici sono importanti per prevedere la prognosi e la gestione della CCAM poiché la presenza di cisti più grandi e/o idrope fetale è considerata ad alto rischio ed è spesso associata a una prognosi peggiore (Dazhi Fan, et al., 2017).
Ecocardiografia fetale per lo studio, oltre che dell'anatomia cardiaca, della "compliance" in caso di compressione da parte del tessuto polmonare anomalo
Consulenza Chirurgo Pediatra
Ulteriori controlli ecografici
I bambini asintomatici dovrebbero essere sottoposti a una TC postnatale anche se la CCAM sembra essersi risolta o diminuita all'ecografia prenatale. Le CCAM hanno una buona prognosi complessiva. Per confermare la risoluzione delle lesioni diagnosticate in epoca prenatale è necessario eseguire una TC piuttosto che la RX Torace (Shamas AG et al., 2017).
Bibliografia
Chen Y, Zhao B, Xi F, Wang Y, Yang M, Luo Q. The prenatal ultrasonic character and postnatal follow-up of 227 microcystic and macrocystic congenital cystic adenomatoid malformations. J Obstet Gynaecol. 2021 May;41(4):562–8.
Crombleholme TM, Beverly Colemann , Holly Hedrick , Kenneth Liechty , Lori Howell , Alan W. Flake , Marco Johnson , N Scott Adzick. Cystic adenomatoid malformation volume ratio predicts outcome in prenatally diagnosed cystic adenomatoid malformation of the lung.J Pediatr Surg. 2002 Mar;37(3):331-8.
Curran PF, Jelin EB, Rand L, Hirose S, Feldstein VA, Goldstein RB, et al. Steroidi prenatali per malformazioni adenomatoidi cistiche congenite microcistiche. J Pediatr Surg. 2010; 45 (1): 145–150.
Dazhi Fan, Shuzhen Wu, Rui Wang, Yi Huang, Yao Fu, Wen Ai, Meng Zeng, Xiaoling Guo, Zhengping Liu. Successfully treated congenital cystic adenomatoid malformation by open fetal surgery: A care-compliant case report of a 5-year follow-up and review of the literature. Medicine (Baltimore) . 2017 Jan;96(2):e5865.
Fetal Medicine Foundation
Leblanc C., Marguerite Baron, Emilie Desselas, Minh Hanh Phan, Alexis Rybak, Guillaume Thouvenin, Clara Lauby, Sabine Irtan. Congenital pulmonary airway malformations: state-of-the-art review for pediatrician's use. Eur J Pediatr . 2017 Dec;176(12):1559-1571.
Macedo Isidora, Lins Parentes Fortes, João Renato Bennini Junior . Use of Corticosteroids in Prenatal Treatment of Congenital Pulmonary Adenomatoid Malformation: Integrative Review. Rev. Bras Ginecol Obstet .2022 marzo;44(3):304-310.
Rolo LC, Ribeiro GD, Caldas JVJ, Coutinho LG, Muniz TD, Araujo, et al. Perinatal outcomes of prenatal diagnosis of congenital pulmonary airway malformation: an experience. Rev Assoc Med Bras. 2022 Nov 28;68(11):1582–6.
Shamas AG, Bohara K. Congenital cystic adenomatoid malformation of the lung (CCAM), a retrospective clinical audit and literature review in a tertiary centre in Scotland over a period of 14 years. J Obstet Gynaecol. 2017;37(1):19-24.
Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Human Pathol. 1977;8(2):155–171.
Tran Hong, Michelle A Fink , Joe Crameri , Fiona Cullinane. Congenital cystic adenomatoid malformation: monitoring the antenatal and short-term neonatal outcome. Aust NZJ Obstet Gynaecol .2008 ottobre;48(5):462-6.
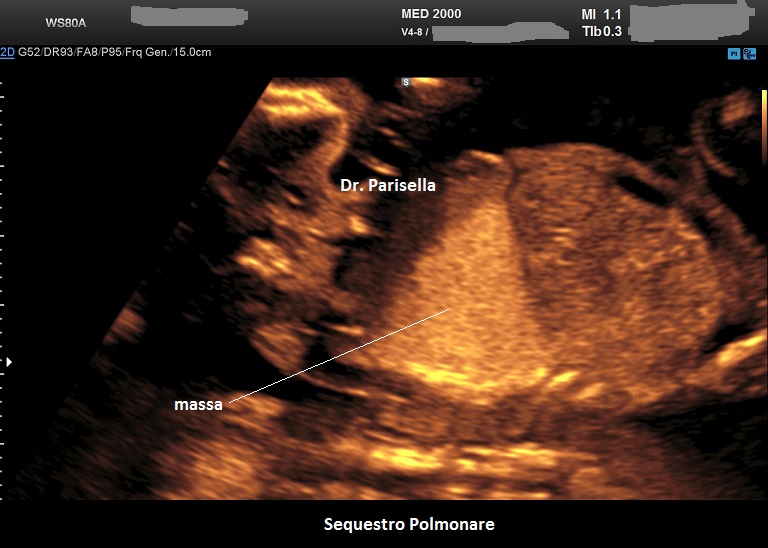
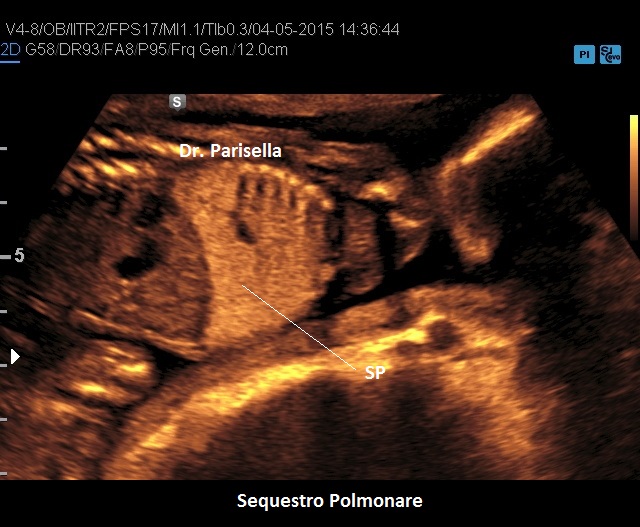
Sequestro Polmonare
Il Sequestro Polmonare è costituito da un'area di parenchima polmonare che non è in comunicazione con l'albero tracheo-bronchiale ed è tributaria della circolazione sistemica e non polmonare. E' una lesione monolaterale, localizzata nella maggior parte dei casi al lobo inferiore del polmone sinistro. Ne esistono due varianti:
- intra-lobare: rappresenta il 75% delle forme diagnosticate in epoca post-natale e quasi mai vengono diagnosticate in utero
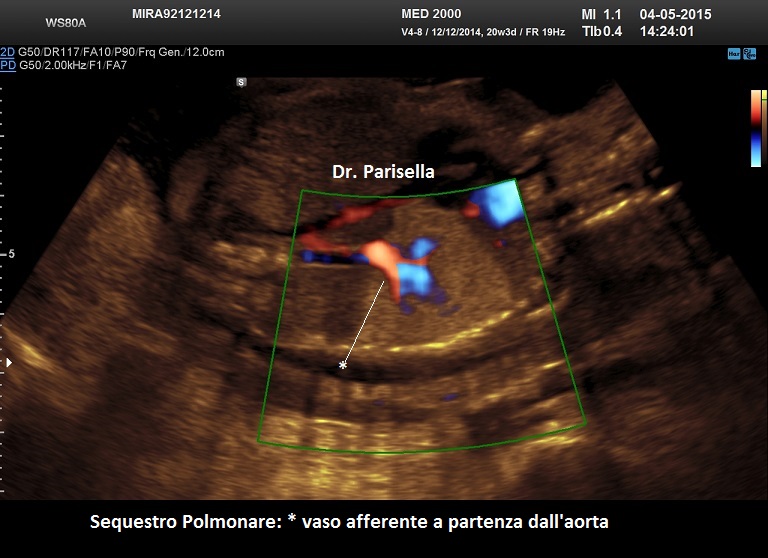
- extra-lobare: è una forma più rara ed è quella classicamente diagnosticata in epoca prenatale; nel 90% dei casi è sopra-diaframmatica, nel 10% è sotto-diaframmatica. Caratteristicamente presenta un vaso afferente a partenza dall'aorta toracica o addominale.
Diagnosi ecografica
Si presenta come una lesione iperecogena localizzata più frequentemente al lobo inferiore del polmone sinistro, più raramente in sede sottodiaframmatica. Al Color-Doppler si evidenzia il vaso afferente a partenza dall'aorta toracica o addominale.
Deve essere valutata la eventuale presenza di idrotorace oppure di idrope. Il sequestro polmonare può associarsi ad altre anomalie toraciche come l'ernia diaframmatica, cisti da duplicazione, fistola tracheo-esofagea, anomalie dell'arteria polmonare
La diagnosi differenziale deve essere posta con la MACP: in questo caso è dirimente il riscontro di un vaso afferente originato dall'aorta nel sequestro polmonare a differenza della MACP in cui la massa è irrorata da diramazioni dell'arteria polmonare.
La prognosi è in genere favorevole in quanto vi è un alto tasso di regressione spontanea o dopo terapia.
I rischi di aneuploidia e sindromico sono bassi.
Management:
Ecocardiografia fetale
Consulenza Chirurgo Pediatra
Ulteriori controlli ecografici
Bibliografia
Bush A, Hogg J, et al. Cystic lung lesions. Prenatal diagnosis and management. Prenatal Diagn, 2008; 28:604-611
Lopoo JB, Goldstein RB, et al. Fetal pulmonary sequestration: a favourable congenital lung lesion. Obst Gyn 1999; 94:567-571
Monni G, Paladini D., et al. Prenatal ultrasound diagnosis of congenital cystic malformation of the lung: a report of 26 cases and review of tra literature. Ultr. Oq Gyn 2000; 16:159-162
Pumbergera W, Houmannb M, et al. Longitudinal observation of antenatally detected congenital lung malformations (CLM): natural history, clinical outcome and long-term follow-up. Eur J Card Surg, 2003; 24:703-711
Aggiornamenti
- Patologie Genetiche dello Scheletro
Sono elencate tutte le 436 Displasie Scheletriche con la... - Consigli per l'utilizzo del software diagnosi in Diagnosi...
Consigli per l'utilizzo del software diagnosi in Diagnosi... - TERMINI DI USO DEL PORTALE WEB med2000eco e Software...
TERMINI DI USO DEL PORTALE WEB med2000eco e Software...