![]()
Acondroplasia - Acondroplasia Eterozigote (OMIM 100800)
Acondroplasia - Acondroplasia Eterozigote OMIM 100800
redatto da Dr. P.Parisella
Il
termine Acondroplasia (FGFR3 chondrodysplasia group) si riferisce alla mancata
formazione della cartilagine di accrescimento delle ossa lunghe degli arti; ha
una incidenza è di circa 1/25000 nati e colpisce indifferentemente i due sessi.
La forma eterozigote è la più comune delle Osteocondrodisplasie (OCD) un gruppo
di circa 200 malattie diverse distinte in base alle caratteristiche cliniche,
genetiche e radiologiche.
Genetica
Il
gene coinvolto nell'acondropasia (ACP) si trova nel cromosoma 4 e contiene
l'informazione per produrre la proteina FGFR3 (recettore del fattore di
crescita dei fibroblasti di tipo 3); questo recettore permette alle cellule di
ricevere un segnale che stimola le cellule a moltiplicarsi. L'ACP è causata da
mutazioni del gene FGFR3 che lo rendono incapace di ricevere il segnale e
trasmetterlo all'interno delle cellule della cartilagine di accrescimento che
come detto necessitano di questo segnale per moltiplicarsi. Il mancato sviluppo
armonico della cartilagine di accrescimento delle ossa lunghe degli arti provoca gravi disturbi della crescita in
lunghezza dell'osso con conseguente nanismo (nanismo acondroplasico).
La
mutazione del gene FGFR3 (G380R) è comune al 99% degli affetti da ACP e circa 9
casi su 10 di pazienti affetti nasce da genitori assolutamente normali. L'ACP
si trasmette con modalità autosomica dominante: una coppia con un solo genitore
affetto ha il 50% di probabilità di avere un figlio affetto; una coppia con
entrambi i genitori affretti ha 1 probabilità su 4 di concepire un figlio
affetto dalla forma più grave (ACP omozigote), 1 probabilità su 2 di avere un
figlio affetto e 1 probabilità su 4 di avere figli normali.
La Diagnosi
prenatale si basa su indagini di laboratorio e su di un esame ecografico
dettagliato.
Diagnosi
di laboratorio
La
diagnosi di laboratorio, in caso di sospetta acondroplasia all'esame ecografico
e/o nel caso in cui uno dei due genitori è affetto da acondroplasia, è
possibile ricercando la mutazione caratteristica del gene FGFR3 mediante test invasivi (villocentesi o
amniocentesi) o mediante analisi del DNA fetale nel sangue materno.
Diagnosi
ecografica
E'
caratterizzata da un accorciamento prevalente del femore (rizomelia), anche se
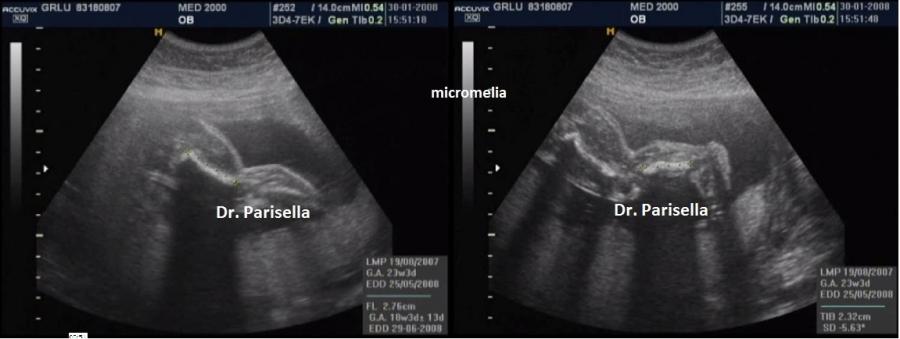
sono interessate tutte le ossa lunghe (micromelia) e le ossa delle mani e dei
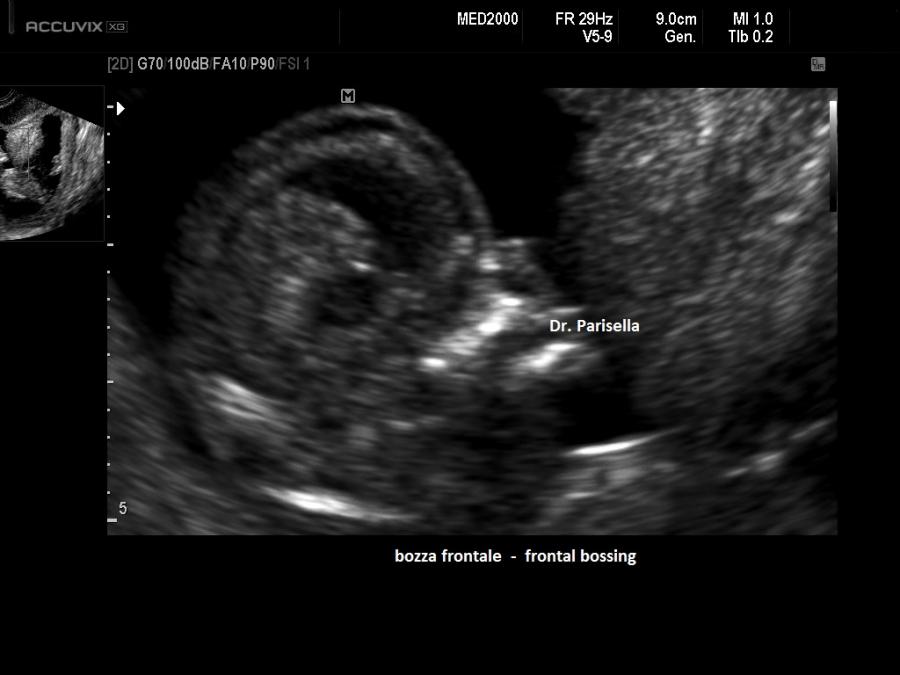
piedi (brachidattilia); la testa è grossa (macrocefalia/macrocrania) con bozze
frontali prominenti e ponte nasale depresso. La diagnosi ecografica precoce non
è possibile. Spesso infatti l'acondroplasia può essere sospettata o confermata
solo in fase avanzata di gravidanza in quanto la micromelia si manifesta
tardivamente con un accrescimento delle ossa lunghe inferiore al 5° percentile
evidente solo nel III trimestre. La diagnosi può essere facilitata in caso di
storia familiare positiva per tale patologia. Alcuni studi hanno riportato
l'associazione con la NT aumentata.
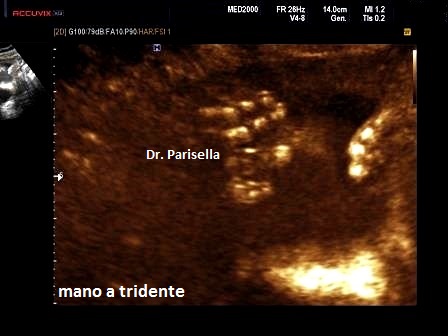
Il
segno ecografico esclusivo (segno principe) da solo sufficiente, se presente, a
porre diagnosi è la Mano a Tridente
(aumento dell'interspazio tra 3° e 4° dito).
Schemattizzando la
diagnosi ecografica si basa sul riscontro di :
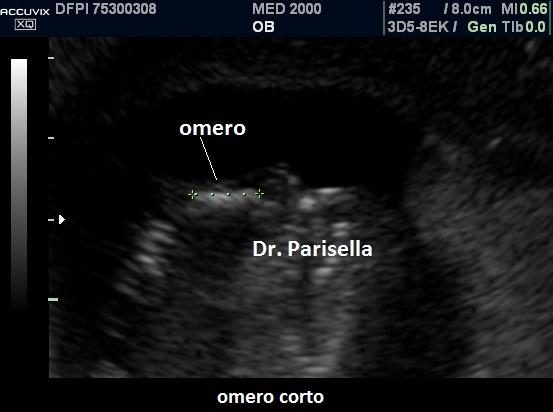
1. micromelia:
è il segno più evidente, con gli arti inferiori al 5°percentile dopo la 20a-22a
settimana; vi è anche brevità rizomelica che interessa principalmente i femori.
2. femore
corto con coscia che presenta un aumentato spessore del sottocute.
3. Bozze frontali
sporgenti
4. Radice
del naso depressa o naso a sella
5. Macrocrania
6. Mano a
tridente (segno esclusivo ma incostante)
7. Ossa di
mani e piedi corte
8. Scoliosi
lombare
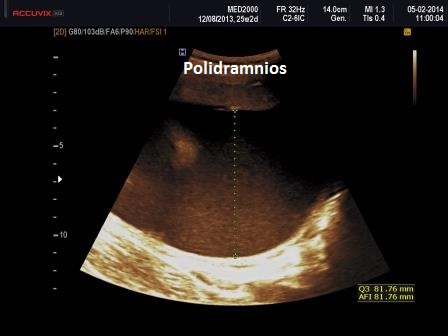
9. Polidramnios
(in fase avanzata di gravidanza)
La Diagnosi
Differenziale si pone con tutte quelle condizioni in cui vi è micromelia e un
femore piccolo:
1)
trisomia 21 eseguendo un cariotipo; le cellule prelevate in corso di
amniocentesi possono essere utilizzate per la ricerca della mutazione alla base
della acondroplasia.
2)
Displasia Campomelica: le ossa sono incurvate e vi è spesso micrognatia.
3)
Displasia Tanatofora tipo I dove la micromelia e il polidramnios sono più
gravi, vi è ipoplasia toracica e non vi è la mano a tridente.
4)
Osteogenesi Imperfetta tipo II dove vi è scarsa mineralizzazione delle ossa,
ossa lunghe di forma angolata per fratture ossee, torace stretto.
5)
Acondrogenesi dove micromelia e polidramnios sono più gravi e vi è scarsissima
mineralizzazione delle ossa.
6)
Displasia Diastrofica dove la micromelia è associata a contratture articolari,
in modo particolare a carico di mani e piedi.
Un
cenno a parte merita la distinzione tra forme eterozigoti ed omozigoti. Uno
studio di Patè et al. ha riportato differenze tra le curve di crescita del
femore nelle forme omozigoti ed eterozigoti.
I feti con acondroplasia omozigote presentavano un accorciamento del
femore al disotto del 3° percentile già a 14 - 17 settimane, mentre i feti con
acondroplasia eterozigote mostravano un accorciamento del femore al disotto del
3° percentile a 18 - 26 settimane.
Bibliografia
Genoma Acondroplasia FGFR3 gene
Bowen, P. Achondroplasia in two sisters with normal parents.
Birth Defects Orig. Art. Ser. X(12): 31-36, 1974
Chitayat, D., Fernandez, B., Gardner, A., Moore, L., Glance,
P., Dunn, M., Chun, K., Sgro, M., Ray, P., Allingham-Hawkins, D. Compound
heterozygosity for the achondroplasia-hypochondroplasia FGFR3 mutations:
prenatal diagnosis and postnatal outcome. Am. J. Med. Genet. 84: 401-405, 1999.
Dennis, J. P., Rosenberg, H. S., Alvord, E. C., Jr.
Megalencephaly, internal hydrocephalus and other neurological aspects of
achondroplasia. Brain 84: 427-445, 1961.
Elejalde, B. R., Elejalde, M. M., Hamilton, P. R., Lombardi,
J. N. Prenatal diagnosis in two pregnancies of an achondroplastic woman. Am. J.
Med. Genet. 15: 437-439, 1983.
Francomano, C. A., Pyeritz, R. E. Achondroplasia is not
caused by mutation in the gene for type II collagen. Am. J. Med. Genet. 29:
955-961, 1988.
Fremion, A. S., Garg, B. P., Kalsbeck, J. Apnea as the sole
manifestation of cord compression in achondroplasia. J. Pediat. 104: 398-401,
1984.
Hall, J. G. The natural history of achondroplasia.In:
Nicoletti, B.; Kopits, S. E.; Ascani, E.; McKusick, V. A. : Human Achondroplasia:
A Multidisciplinary Approach. New York: Plenum Press (pub.) 1988. Pp. 3-10.
Henderson, S., Sillence, D., Loughlin, J., Bennetts, B.,
Sykes, B. Germline and somatic mosaicism in achondroplasia. J. Med. Genet. 37:
956-958, 2000.
Huggins, M. J., Smith, J. R., Chun, K., Ray, P. N., Shah, J.
K., Whelan, D. T. Achondroplasia-hypochondroplasia complex in a newborn infant.
Am. J. Med. Genet. 84: 396-400, 1999.
Hunter, A. G. W., Bankier, A., Rogers, J. G., Sillence, D.,
Scott, C. I., Jr. Medical complications of achondroplasia: a multicentre
patient review. J. Med. Genet. 35: 705-712, 1998.
Langer, L. O., Jr., Schaefer, G. B., Wadsworth, D. T.
Patient with double heterozygosity for achondroplasia and pseudoachondroplasia,
with comments on these conditions and the relationship between
pseudoachondroplasia and multiple epiphyseal dysplasia, Fairbank type. Am. J.
Med. Genet. 47: 772-781, 1993.
Nicoletti, B., Kopits, S. E., Ascani, E., McKusick, V. A.
Human Achondroplasia: A Multidisciplinary Approach. New York: Plenum Press
(pub.) 1988. Pp. 3-9.
Sobetzko, D., Braga, S., Rudeberg, A., Superti-Furga, A.
Achondroplasia with the FGFR3 1138g-a (G380R) mutation in two sibs sharing a 4p
haplotype derived from their unaffected father. (Letter) J. Med. Genet. 37:
958-959, 2000.
Sommer, A., Young-Wee, T., Frye, T.
Achondroplasia-hypochondroplasia complex. Am. J. Med. Genet. 26: 949-957, 1987.
Aterman, K., Welch, J. P., Taylor, P. G. Presumed homozygous achondroplasia: a review and report of a further case. Path. Res. Pract. 178: 27-39, 1983.
_j5uc5592.jpg)

Aggiornamenti
- Patologie Genetiche dello Scheletro
Sono elencate tutte le 436 Displasie Scheletriche con la... - Consigli per l'utilizzo del software diagnosi in Diagnosi...
Consigli per l'utilizzo del software diagnosi in Diagnosi... - TERMINI DI USO DEL PORTALE WEB med2000eco e Software...
TERMINI DI USO DEL PORTALE WEB med2000eco e Software...