![]()
Sindrome di Edwards o Trisomia 18
Sindrome di Edwards o Trisomia 18
Redatto da: P.ParisellaLa Sindrome di Edwards o Trisomia 18 è una anomalia cromosomica caratterizzata da un cromosoma 18 in sovrannumero. Ha una incidenza di circa 1/6000-1/8000 nati ma l'incidenza è molto più alta (1/2500-1/2600) se si considera che oltre il 95% di feti affetti muore in utero o perchè la gravidanza viene interrotta per la diagnosi prenatale. E' la trisomia più comune dopo la trisomia 21; la prevalenza aumenta con l'età materna e il rischio di ricorrenza in caso di precedente feto affetto da trisomia 21 è di circa 1%.
Il meccanismo che è più frequentemente causa della trisomia 18 è la
non disgiunzione dei cromosomi durante il processo di divisione cellulare
all'interno del gamete di uno dei genitori; prima del concepimento l'ovulo o lo
spermatozoo conservano entrambe le copie del cromosoma 18 all'interno della
cellula; quando avviene la fecondazione, con l'apporto paterno o materno di
un'altro cromosoma 18, si formano cellule trisomiche cioè con tre copie del
cromosoma 18. La maggiore frequenza della trisomia 18 con l'aumentare dell'età
materna sembra legata proprio al fatto che con l'aumentare dell'età materna
sono più frequenti i fenomeni di non disgiunzione.
Le caratteristiche più comuni sono:
- difetto di crescita prenatale e postnatale
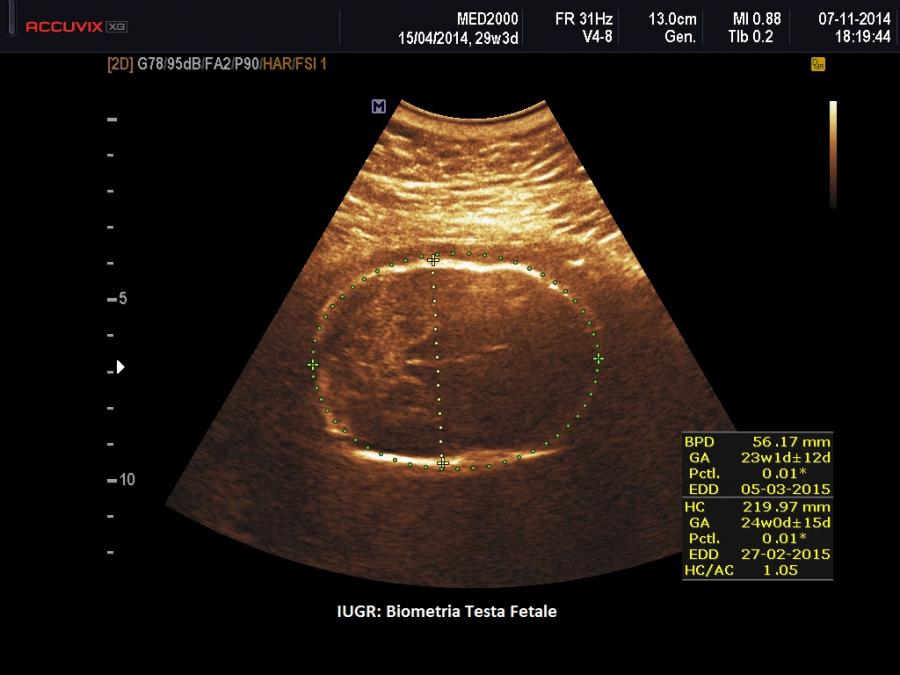
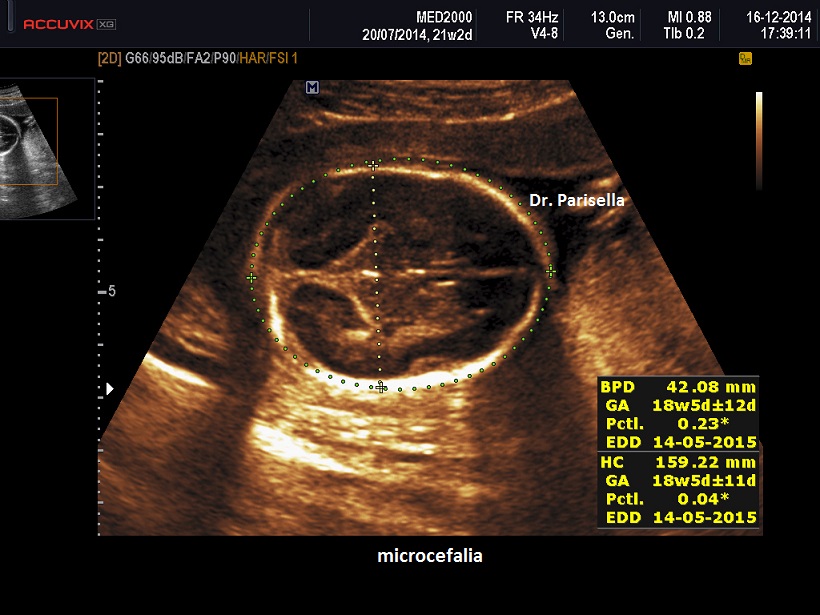
- microcefalia con dolicocefalia
- microretrognazia
- ipertelorismo
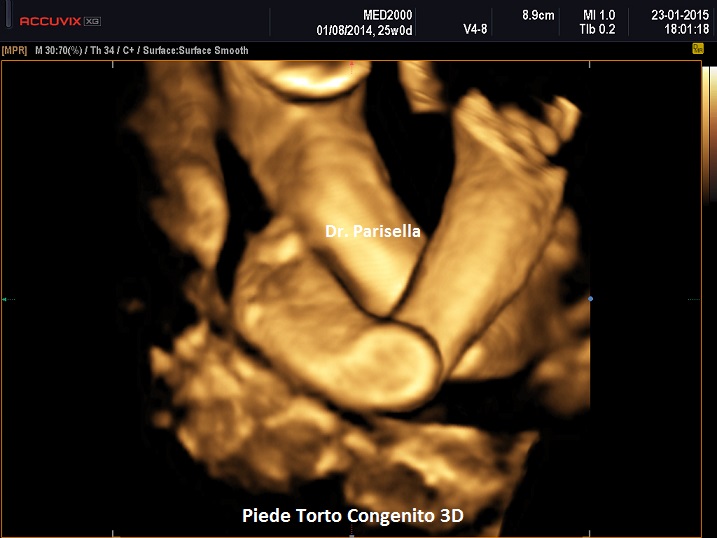
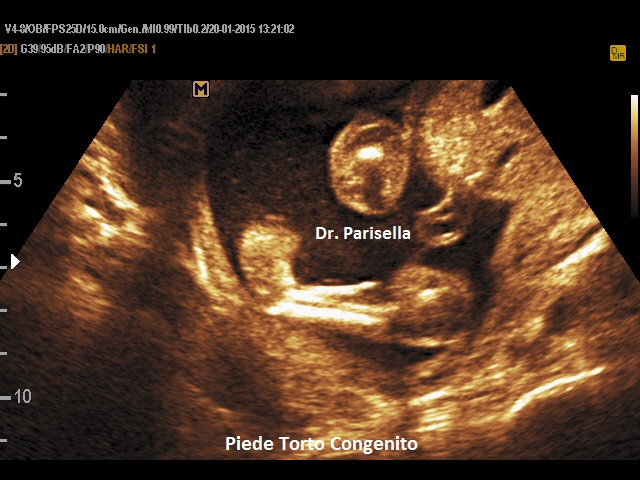
- piede torto congenito (piede equino varo).
Frequenti le malformazioni di vari organi ed apparati:
- anomalie cardiache: presenti nel 80% dei casi
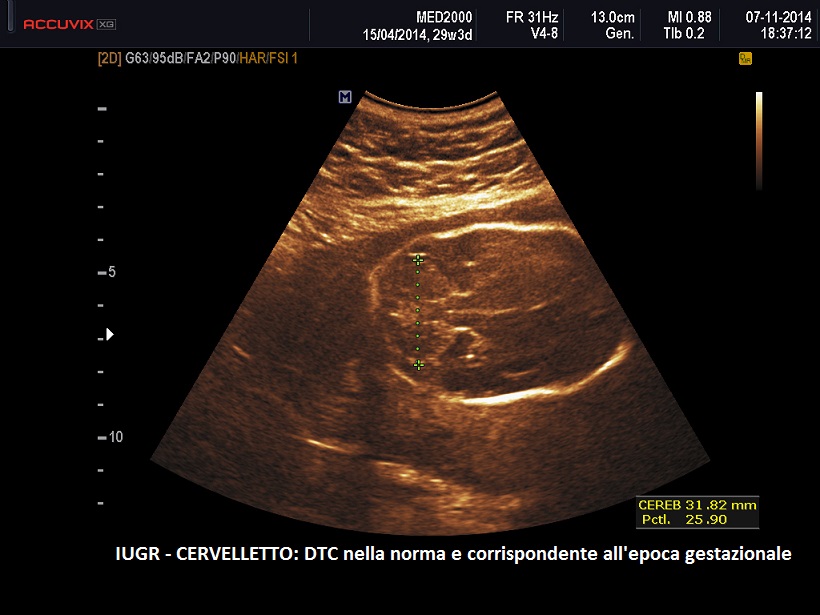
- anomalie del SNC: presenti nel 90% dei casi > agenesia del corpo calloso, idroceflia
- anomalie oculari: microftalmia, coloboma
- anomalie del tubo digerente: atresia esofagea, malformazioni anorettali
- anomalie genito-urinarie: idronefrosi, agenesia renale mono-bilaterale
Meno frequenti sono:
- labiopalatoschisi
- artrogriposi
- aplasia del radio
- spina bifida
- anencefalia
- oloprosencefalia
- onfalocele
- polidattilia
- ernia ombelicale
I segni ecografici più frequenti sono:
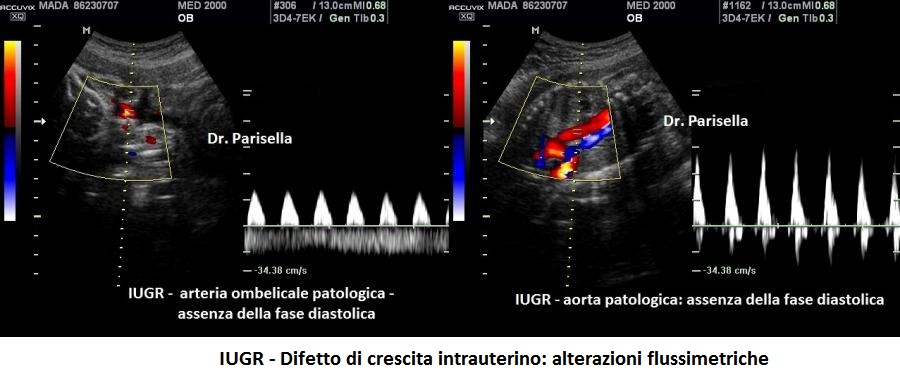
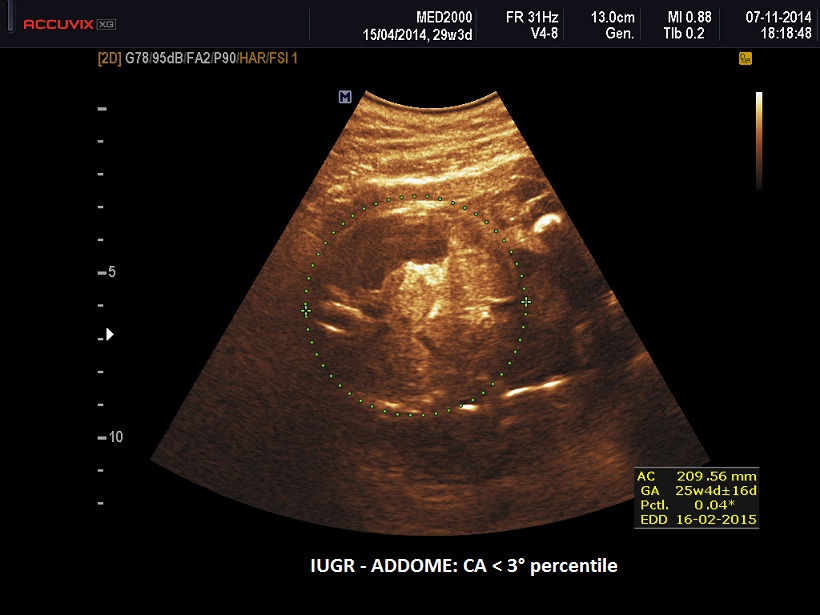
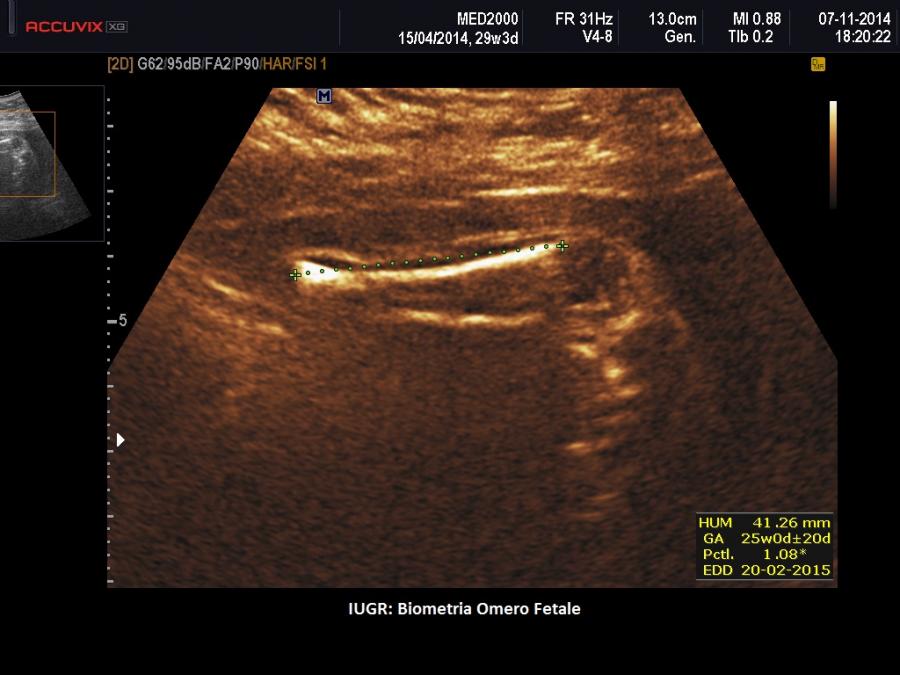
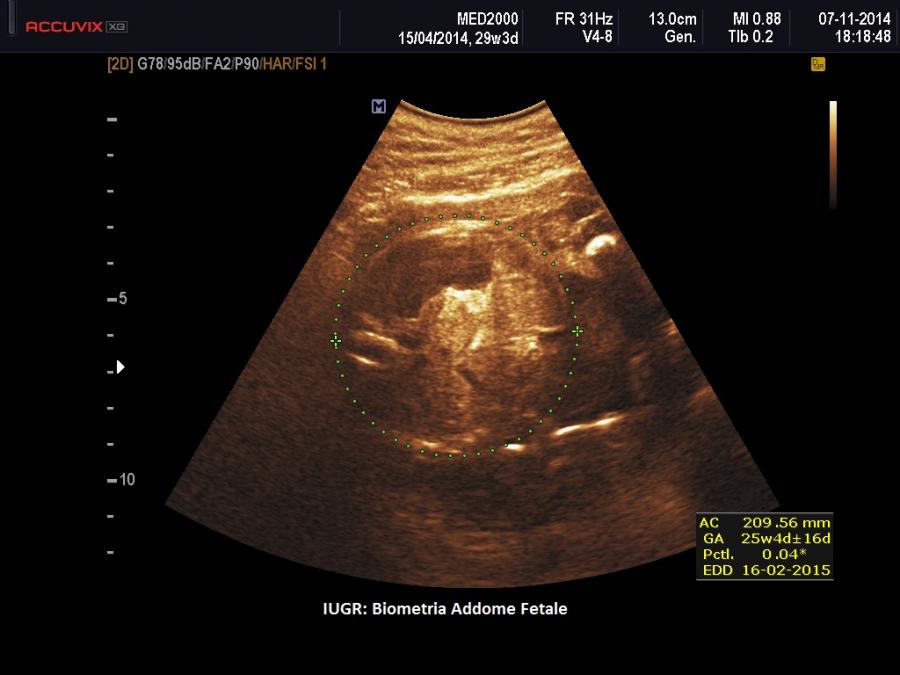
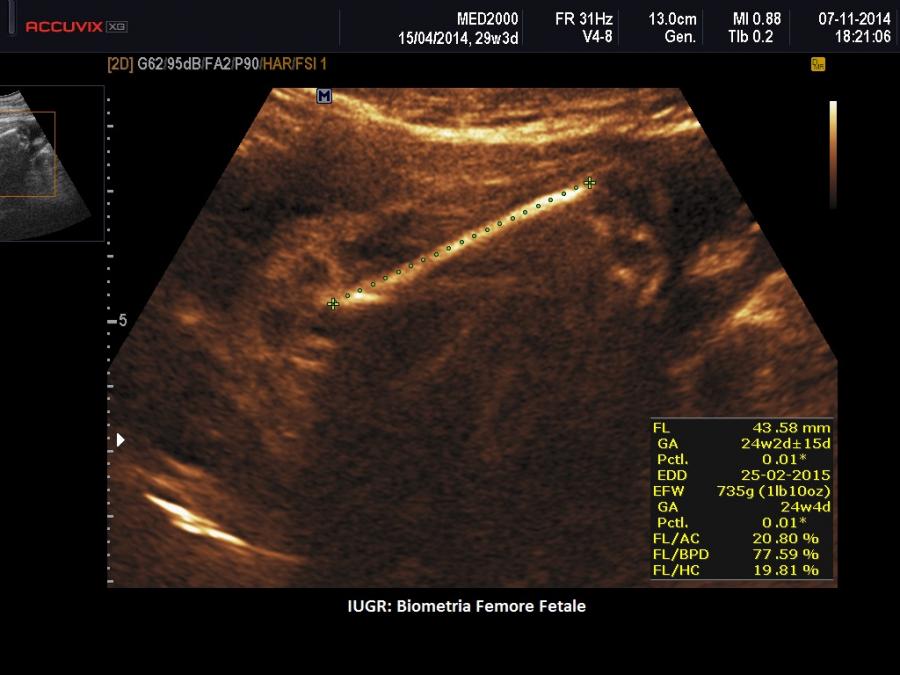
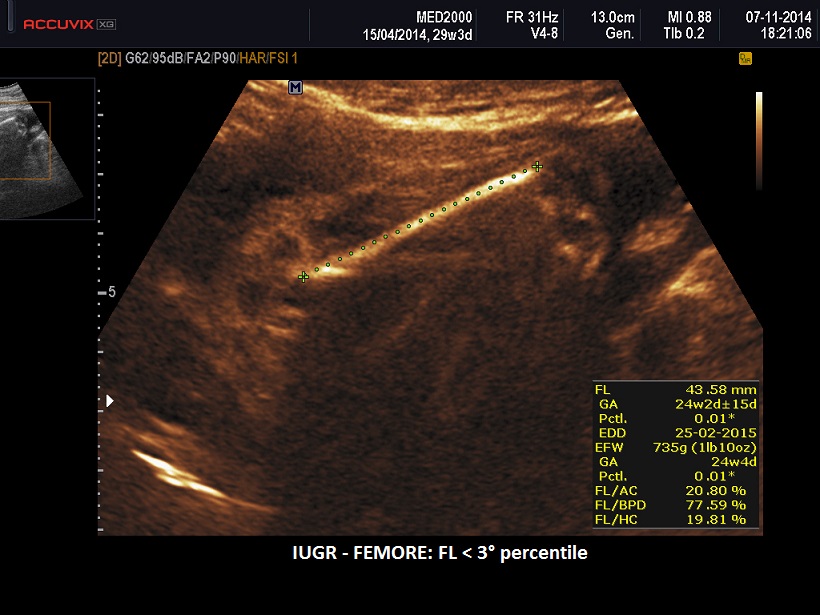
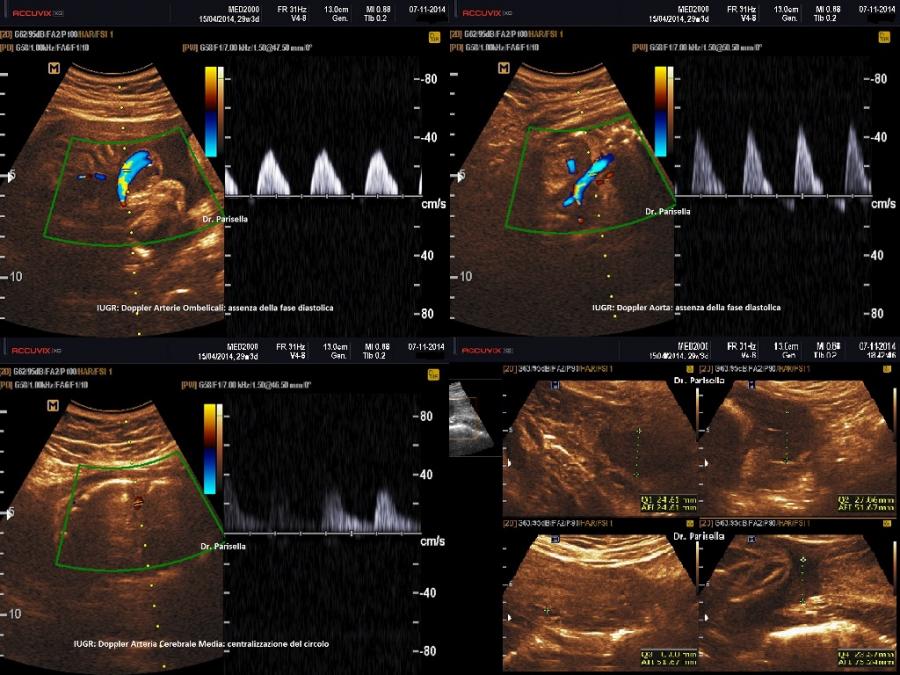
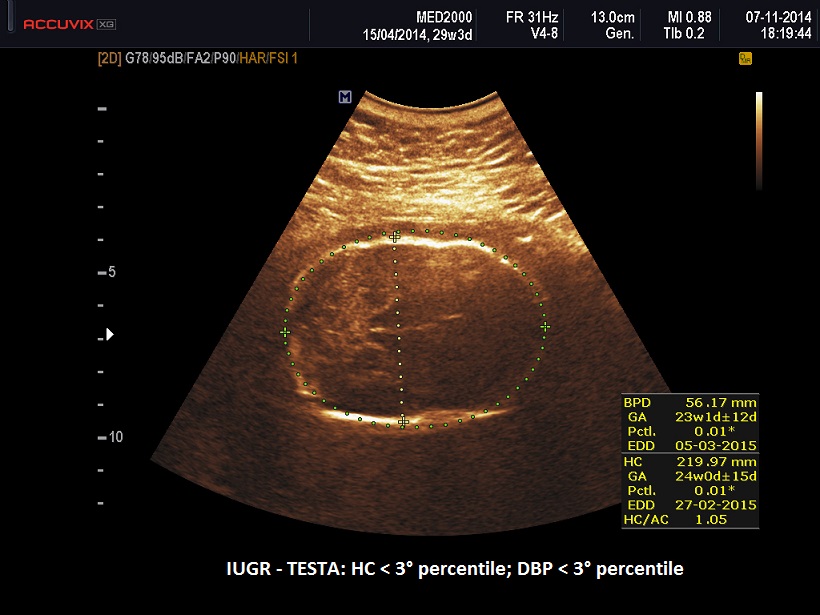
- IUGR
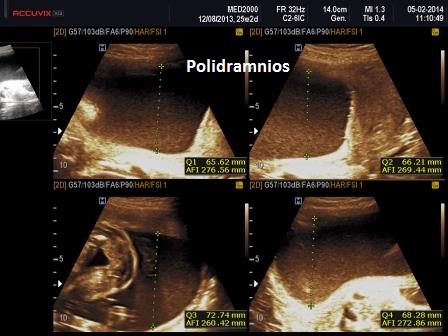
- polidramnios
- cisti dei plessi corioidei
- igroma cistico
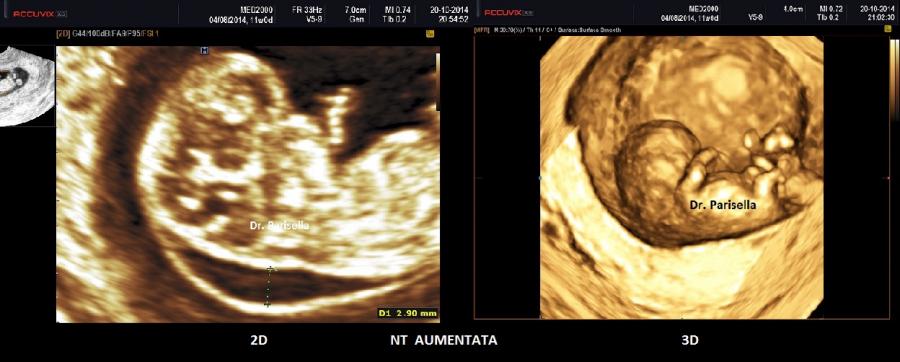
- NT ispessita
- piedi torti.
Altamente diagnostiche sono alcune associazioni:
- cisti dei plessi corioidei, IUGR, piedi torti;
- golf ball, ventricolo sinistro ipoplasico.
Nel I trimestre i segni ecografici possono essere:
- NT ispessita
- arteria ombelicale unica
- assenza dell'osso nasale
- ritardo di crescita precoce
- onfalocele (può costituire un marcatore precoce della sindrome)
Dal punto di vista biochimico vi è una diminuzione della beta-hCG e della PAPP-A.
Vi sono forme caratterizzate da mosaicismo in cui il quadro clinico può variare da una forma fenotipica classica a forme con fenotipo normale in rapporto al numero di cellule con trisomia presenti nei tessuti.
Criteri diagnostici
Ecografici:
I trimestre: NT ispessita, Arteria Ombelicale Unica, assenza dell'osso nasale, onfalocele, IUGR precoce
II trimestre: IUGR, Cisti dei plessi corioidei, Anomalie strutturali (piede torto congenito)
Biochimici: diminuzione beta-hCG, diminuzione PAPP-A
Genetici: cariotipo con cromosoma sovrannumerario nella regione 18q11-q12.

Fenotipo
Basso peso alla nascita, cranio dolicocefalico, orecchie malconformate "a fauno" (elice appuntito) e ad impianto basso. Ipoplasia della mandibola, mani flesse con indice sovrapposto al medio, solco palmare unico, piede equino-varo con tallone prominente (piede a picozza). Frequenti sono le malformazioni cardiache, delle vie urinarie e dell'apparato digerente.
Il 30% dei pazienti muore entro il primo mese di vita per le gravi anomalie viscerali e solo il 10% sopravvive oltre l'anno di vita con un severo ritardo di sviluppo psicomotorio.
Bibliografia
Aziz AM. Muscular and other abnormalities in a case of Edward?s syndrome (18-trisomy). Teratology 1979; 20: 303-312.
Bersu ET, Ramirez-Castro JL. Anatomical analysis of the developmental effects of aneuploidy in man - the 18 trisomy syndrome. Syndromes of the head and neck. Am J Med Genet 1977; 1: 173-193.
Crider KS, Olney RS, Cragan JD. Trisomies 13 and 18: Population prevalences, characteristics, and prenatal diagnosis, metropolitan Atlanta 1994-2003. Am J Med Genet A 2008; 146: 820-826.
De Souza E, Halliday J, Chan A, Bower C, Morris JK. Recurrence risks for trisomies 13, 18 and 21. Am J Med Genet A 2009; 149: 16-22.
Geiser CF. Long survival in a male with 18-trisomy syndrome and Wilm?s tumour: a subsequent report. Pediatrics 1969; 44: 153.
Goldstein H, Nielsen KJ. Rates and survival of individuals with trisomy 13 and 18. Clin Genet 1988; 34: 366-372.
Hodes ME, Cole J, Palmer G, Reed T. Clinical experience with trisomy 18 and 13. J Med Genet 1978; 15: 48-60.
Kaneko Y, Kobayasihi J, Ymamoto Y, Yoda H, Kanetaka Y, Nakajima Y et al. Intensive cardiac management in patients with trisomy 13 or trisomy 18. Am J Med Genet A 2008; 146: 1372-1380.
Michaelson PS, Gilles FH. Central nervous system abnormalities in trisomy 18 syndrome. J Neurol Sci 1972; 15: 193-208.
Smith A, Field B, Learoyd BM. Trisomy 18 at age 21 years. Am J Med Genet 1989; 23: 256-257.
Urban B, Bersu ET. Chromosome 18 aneuploidy: Anatomical variations observed in cases of full and mosaic trisomy 18 and a case of deletion of the short arm of chromosome 18. Am J Med Genet 1987; 27: 425-434.
Young ID, Cook JP, Mehta L. Changing demography of trisomy 18. Arch Dis Child 1986; 61: 1035-1036.

Aggiornamenti
- Patologie Genetiche dello Scheletro
Sono elencate tutte le 436 Displasie Scheletriche con la... - Consigli per l'utilizzo del software diagnosi in Diagnosi...
Consigli per l'utilizzo del software diagnosi in Diagnosi... - TERMINI DI USO DEL PORTALE WEB med2000eco e Software...
TERMINI DI USO DEL PORTALE WEB med2000eco e Software...